POSS基丙烯酰氧基超支化聚乙烯及其在石墨烯/碳纳米管杂化电热涂层中的应用
poss基丙烯酰氧基超支化聚乙烯及其在石墨烯/碳纳米管杂化电热涂层中的应用
技术领域
1.本发明涉及一种poss基丙烯酰氧基超支化聚乙烯及其在石墨烯/碳纳米管杂化电热涂层中的应用。
背景技术:
2.碳基电热材料由于其优异的电热性能、电热转化效率和升温速率受到广泛关注,在能源材料、生命健康、光电信息等领域具有广阔的应用前景。但是碳基电热材料的应用过程中仍然存在缺乏有效稳定性、材料耐热温度过低、容易引起安全事故等问题。
3.为了解决稳定性,一系列关于碳基电热材料的应用方法已经得到了报道,其中最主要应用方法就是添加工程类塑料作为基体对碳材料进行分散。借助此方法所获得的电热材料具有较高的稳定性,然而工程类塑料普遍不具备导电、导热能力,填充会导致塑料电热性能的大量损失,使其丧失电热性能。
4.结合前期研究过程中的技术积累所知在合适的有机溶剂如四氢呋喃或氯仿中,利用超支化聚乙烯与碳材料表面的非共价ch-π作用,借助超声可有效剥离碳材料,获得浓度较高的碳材料有机分散液,同时超支化聚乙烯在溶剂中能够作为稳定剂分散碳材料方便后续的应用。
5.引入丙烯酰胺异丁基倍半硅氧烷(poss)和1,4-丁二醇二丙烯酸酯两种单体参与合成超支化聚乙烯——poss基丙烯酰氧基超支化聚乙烯。利用这种超支化聚合物,在合适的有机溶剂如四氢呋喃或氯仿中,利用超支化聚合物与石墨烯表面的非共价ch-π作用,借助超声可有效剥开天然石墨,获得浓度较高的石墨烯有机分散液时所得石墨烯结构缺陷少,分散稳定;利用超支化聚合物分散碳纳米管得到高浓度的碳纳米管分散液。poss基丙烯酰氧基超支化聚乙烯可由催化剂α-二亚胺钯(pd-diimine)催化乙烯聚合,以一步法“链移走”机理获得。另外,在超支化聚乙烯合成中引入poss基团提供了晶区结构有利于提升涂层的升温速率并且它所特有的无机硅氧骨架对于涂层的耐热性能具有提升作用;而引入的丙烯酰氧基团则由于其具有的光固化能力,对于涂层的稳定性具有较大的提升能力。
技术实现要素:
6.本发明的第一个目的是提供poss基丙烯酰氧基超支化聚乙烯(hbpe@acryl@poss)。
7.本发明的第二个目的是提供hbpe@acryl@poss在制备高稳固且耐燃的石墨烯/碳纳米管杂化电热涂层中的应用,该涂层获得了更好的升温速率、耐弯折性能和耐热性能。
8.下面对本发明采用的技术方案做具体说明。
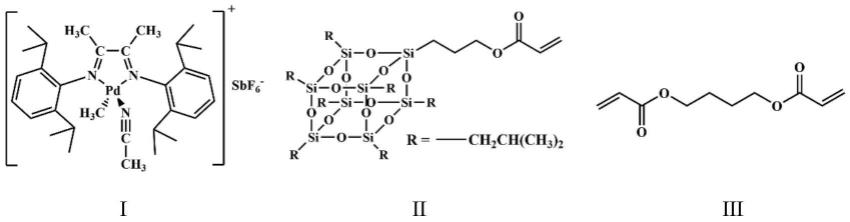
9.第一方面,本发明提供一种poss基丙烯酰氧基超支化聚乙烯(hbpe@acryl@poss),其由式i所示的pd-diimine催化剂催化乙烯、式ii所示的丙烯酰胺异丁基倍半硅氧烷单体(poss)和式iii所示的1,4-丁二醇二丙烯酸酯单体以一步法“链移走”共聚机理聚合获得;
[0010][0011]
其中r为异丁基。
[0012]
作为优选,所述poss基丙烯酰氧基超支化聚乙烯的具体制备方法如下:在乙烯保护下往反应容器中加入丙烯酰胺异丁基倍半硅氧烷单体、1,4-丁二醇二丙烯酸酯单体和无水级溶剂,并控制温度在15~35℃,然后加入溶于无水级溶剂中的pd-diimine催化剂,在温度15~35℃、乙烯压力0.1~6atm的条件下搅拌反应12~48小时,聚合结束后将所得产物倒入酸化甲醇中使聚合终止,所得聚合反应混合物经分离纯化得到poss基丙烯酰氧基超支化聚乙烯。
[0013]
作为进一步的优选,所述无水级溶剂选自下列之一:无水二氯甲烷、无水三氯甲烷、无水甲苯。
[0014]
作为进一步的优选,所述聚合反应体系中,丙烯酰胺异丁基倍半硅氧烷单体的初始浓度为0.3~0.6mol/l,1,4-丁二醇二丙烯酸酯单体的初始浓度为0.08~0.70mol/l,pd-diimine催化剂的初始浓度为10~12.5mg/ml。
[0015]
作为进一步的优选,所述聚合反应体系中,丙烯酰胺异丁基倍半硅氧烷单体与1,4-丁二醇二丙烯酸酯单体的投料摩尔比为7.5:1~1:1,更进一步优选为7.5:1~4:1。
[0016]
作为进一步的优选,反应温度为30℃,乙烯压力为0.1~1atm,反应时间为24h。
[0017]
作为进一步的优选,聚合反应混合物的分离纯化按照如下步骤进行:
[0018]
(a)聚合反应混合物先除去溶剂;
[0019]
(b)将所得的产物溶解于四氢呋喃,加入少量的双氧水和浓盐酸搅拌1~5小时溶解聚合物中的钯颗粒,随后用甲醇进行沉淀并除去溶剂;
[0020]
(c)将所得的产物溶解于四氢呋喃,利用甲醇进行聚合物的沉淀去除游离的1,4-丁二醇二丙烯酸酯单体,此过程重复2~3遍;
[0021]
(d)将所得的产物溶解于甲苯,利用甲醇进行聚合物的沉淀去除游离的丙烯酰胺异丁基倍半硅氧烷单体,此过程重复2~3遍;
[0022]
(e)所得产物经30~50℃下真空干燥24~48h后分别获得poss基丙烯酰氧基超支化聚乙烯。
[0023]
第二方面,本发明提供了poss基丙烯酰氧基超支化聚乙烯在制备石墨烯/碳纳米管杂化电热涂层中的应用,具体应用方法包括如下步骤:
[0024]
(1)将石墨粉、poss基丙烯酰氧基超支化聚乙烯与有机溶剂a进行混合,经密封后对所得混合物进行超声,获得石墨烯初始分散液b,进一步经低速离心及静置处理获得含有过量poss基丙烯酰氧基超支化聚乙烯的石墨烯分散液c,对所得石墨烯分散液c进行高速离心和/或真空抽滤去除游离的poss基丙烯酰氧基超支化聚乙烯,再次超声使之分散到有机溶剂a后,获得石墨烯分散液;其中,石墨粉的投料浓度为0.1~1500mg/ml,poss基丙烯酰氧基超支化聚乙烯与石墨粉的质量比为0.005~10:1;
[0025]
(2)将碳纳米管粉末、poss基丙烯酰氧基超支化聚乙烯与有机溶剂d进行混合,经密封后对所得混合物进行超声,获得碳纳米管初始分散液e,进一步经低速离心及静置处理获得含有过量poss基丙烯酰氧基超支化聚乙烯的碳纳米管分散液f,对所得碳纳米管分散液f进行高速离心和/或真空抽滤去除所含的过量poss基丙烯酰氧基超支化聚乙烯,再次超声使之分散到有机溶剂d后,获得稳定分散的碳纳米管分散液;其中,碳纳米管的投料浓度为0.1~1500mg/ml,poss基丙烯酰氧基超支化聚乙烯与碳纳米管的质量比为0.005~10:1;
[0026]
(3)将步骤(1)制得的石墨烯分散液、步骤(2)制得的碳纳米管分散液和2,2-二甲氧基-2-苯基苯乙酮混合均匀,随后利用真空抽滤的方式将混合液抽滤在孔径为0.1~0.2μm的聚四氟乙烯过滤膜上,然后待膜层中的溶剂自然挥发后得到固态膜,在200~1500w的紫外灯下固化3~20分钟,形成一张稳定的电热涂层;其中石墨烯分散液与碳纳米管分散液按照石墨烯与碳纳米管的体积比为5:1~1:5投料,所述2,2-二甲氧基-2-苯基苯乙酮的投料浓度为0.1~3mg/ml。
[0027]
本发明步骤(1)所述的石墨粉可采用如下来源之一:天然磷片状石墨或膨胀石墨,优选天然磷片状石墨;所述石墨粉的颗粒尺寸控制于50~1500目范围之间。
[0028]
本发明步骤(1)所述的有机溶剂a可采用如下分析纯或化学纯溶剂之一:三氯甲烷、四氢呋喃、氯苯、正庚烷、二氯甲烷。
[0029]
本发明步骤(1)中,石墨粉的投料浓度优选为0.5~500mg/ml,poss基丙烯酰氧基超支化聚乙烯与石墨粉投料质量比为0.1~10:1。
[0030]
本发明步骤(1)中,所述的超声推荐在超声功率为20~300w、恒温15~35℃的条件下进行,持续超声时间优选为12~150h,以获得石墨烯初始分散液b。所述的低速离心推荐在室温、2000~5000rpm的条件下进行,离心时间优选为25~60min。所述的静置处理时间优选为1~4h。
[0031]
本发明所述的碳纳米管可采用如下来源之一:单壁碳纳米管、双壁碳纳米管、多壁碳纳米管、高导电多壁碳纳米管、羟基化碳纳米管、羧基化碳纳米管和氨基化碳纳米管组成的组中的至少一种;所述碳纳米管的平均长度尺寸控制于5-100nm范围之间,优选20nm的多壁碳纳米管。
[0032]
本发明步骤(2)中,所述的有机溶剂d选自下列化学纯或分析纯的试剂之一:四氢呋喃、氯仿、氯苯、二氯甲烷、甲苯、正庚烷。
[0033]
本发明步骤(2)中,碳纳米管的投料浓度优选为0.5~500mg/ml,hbpe@acryl@poss与碳纳米管的投料质量比为0.1~10:1。
[0034]
本发明步骤(2)中,所述的超声推荐在超声功率为20~300w、恒温15~35℃的条件下进行,持续超声时间优选为12~150h,以获得碳纳米管初始分散液e。所述的低速离心推荐在室温、2000~5000rpm的条件下进行,离心时间优选为25~60min。所述的静置处理时间优选为1~4h。
[0035]
本发明相对于现有技术具有如下突出的优点和有益效果:
[0036]
第一,利用pd-diimine催化剂催化乙烯和两种单体(丙烯酰胺异丁基倍半硅氧烷(poss)和1,4-丁二醇二丙烯酸酯)进行聚合合成了一种新的功能性超支化聚合物。
[0037]
第二,利用相对简单的方式制备了可uv固化的高稳固柔性电热涂层。并且制备所得涂层具有较低的方阻、较高的电热温度和耐温性能、较高的电压承载能力、较快的升降温
速率、较好的耐弯折性能。
附图说明
[0038]
图1是本发明合成hbpe@acryl@poss和hbpe@acryl的一种具体实施方式的示意图;
[0039]
图2的(a)和(b)分别是实施例1制备的hbpe@acryl@poss和比较例1制备的的hbpe@acryl的1h核磁共振波谱图、凝胶渗透色谱图和实物图;
[0040]
图3a、3b和3c分别实施例1制备的hbpe@acryl@poss和比较例1制备的的hbpe@acryl的傅里叶红外光谱、x射线衍射图和热重分析图;
[0041]
图4a、4b和4c分别是实施例2的hbpe@acryl@poss基碳纳米管分散液和hbpe@acryl@poss基碳纳米管分散液的热重图、比较例2的hbpe@acryl基石墨烯分散液和hbpe@acryl基碳纳米管分散液的热重图、实施例2的hbpe@acryl@poss基碳纳米管分散液与比较例2的hbpe@acryl基石墨烯分散液的x射线衍射图比较;
[0042]
图5是本发明制备可紫外光固化的柔性电热涂层的示意图;
[0043]
图6:(a)实施例3与比较例3制备的电热涂层的方块电阻阻值比较;(b)实施例3与比较例3制备的电热涂层在不同电压条件下电热温度的比较;(c)实施例3与比较例3制备的电热涂层的电热温度的图像;(d)实施例3与比较例3制备的电热涂层的sem微观图;
[0044]
图7:(a)实施例3制备的电热涂层的升温、降温散点图;(b)比较例3制备的电热涂层的升温、降温散点图;(c)实施例3的hbpe@acryl@poss基电热涂层与比较例3的hbpe@acryl基电热涂层0~60℃动态比热分析图;
[0045]
图8是本发明碳基电热涂层的弯折示意图;
[0046]
图9:(a)实施例4的hbpe@acryl@poss基电热涂层和比较例4的hbpe@acryl基电热涂层初始、弯折数码照片;(b)实施例4的hbpe@acryl@poss基电热涂层和比较例4的hbpe@acryl基电热涂层在不同弯折次数下的电热温度;
[0047]
图10是实施例5制备的的hbpe@acryl@poss和比较例5制备的hbpe@acryl的1h核磁共振波谱图、凝胶渗透色谱图和实物图;
[0048]
图11:(a)实施例6制备的电热涂层的热重图,(b)比较例6制备的电热涂层的热重图;
[0049]
图12:(a)实施例7与比较例7制备的电热涂层的方块电阻阻值比较;(b)实施例7与比较例7制备的电热涂层在不同电压条件下电热温度的比较;(c)实施例7与比较例7制备的电热涂层的电热温度的图像。
具体实施方式
[0050]
下面结合具体实施例和附图对本发明做进一步详细的描述,但本发明的实施方式并不仅限于此。
[0051]
实施例1、比较例1
[0052]
1、样品的制备
[0053]
(1)实施例1样品的制备按如下步骤进行:
[0054]
第1步:在乙烯气体(0.1mpa)的保护下,在250ml的schlenk瓶中依次加入20g poss单体(0.864mol/l)和3g 1,4-丁二醇二丙烯酸酯单体(0.576mol/l),随后加入pd-diimine
催化剂200mg溶解于10ml无水二氯甲烷当中,利用油浴锅控制反应的温度在30℃,随后加入10ml无水二氯甲烷在600r/min的搅拌下反应24h。聚合反应24h后,将所得到的产物转移到200ml的酸化甲醇当中,用于终止聚合反应。
[0055]
第2步:将所得的聚合产物进行纯化,具体操作如下:在常温条件下吹除溶剂,随后分别滴加20滴双氧水和稀盐酸并加入50ml四氢呋喃搅拌2h去除聚合物中的pd颗粒;随后加入100ml甲醇使聚合产物沉淀析出。随后再加入20ml四氢呋喃对产物进行溶解,再用甲醇重新沉淀,该步骤重复2~3次;加入20ml甲苯对产物进行溶解,再用甲醇重新沉淀,该步骤重复2~3次,所得产物经30℃下真空干燥48h后获得hbpe@acryl@poss。该实施例制备的hbpe@acryl@poss供后续实施例使用。
[0056]
(2)比较例1样品的制备按如下步骤进行:
[0057]
第1步:在乙烯气体的保护下,在250ml的schlenk瓶中加入3g 1,4-丁二醇二丙烯酸酯单体(0.576mol/l),随后加入pd-diimine催化剂200mg溶解于10ml无水二氯甲烷当中,利用油浴锅控制反应的温度在30℃,随后加入10ml无水二氯甲烷在600r/min的搅拌下反应24h。聚合反应24h后,将所得到的产物转移到200ml的酸化甲醇当中,用于终止聚合反应。
[0058]
第2步:将所得的聚合产物进行纯化,具体操作如下:在常温条件下吹除溶剂,随后分别滴加20滴双氧水和稀盐酸并加入50ml四氢呋喃搅拌2h去除聚合物中的pd颗粒;随后加入100ml甲醇使聚合产物沉淀析出。随后再加入20ml四氢呋喃对产物进行溶解,再用甲醇重新沉淀,该步骤重复2~3次得到最终产物,所得产物经30℃下真空干燥48h后获得hbpe@acryl。
[0059]
2、表征与测试
[0060]
(1)1h核磁共振波谱测试
[0061]
hbpe@acryl@poss和hbpe@acryl的1h核磁共振波谱(1h nmr)由500mhz anance iii型核磁共振仪(瑞士bruker公司)测得,溶剂为氘代氯仿,测试温度为室温。
[0062]
(2)凝胶渗透色谱分析
[0063]
测试仪器为英国malvern公司生产的omnisec型凝胶渗透色谱仪。测试温度为30℃,ps为标样,thf为流动相。样品制备:聚合物配置成浓度为5mg
·
ml-1
的thf溶液,体积6ml左右,用孔径为0.45微米的有机膜过滤后用于测试。
[0064]
(3)x射线衍射分析
[0065]
测试仪器为荷兰pnalytical公司生产的x pert pro型x射线衍射分析仪。x射线源为cu靶kα射线测试电压40kv,电流40ma,扫描速度10
°
/min,扫描范围5
–
80
°
。聚合物分散液滴在玻璃片上,溶剂充分干燥后直接用于测试。
[0066]
(4)傅里叶红外光谱分析
[0067]
测试仪器为美国nicolet公司生产的nicolet型傅立叶红外光谱议。测试范围在400~4000cm-1
,分辨率为4cm-1
,扫描次数32次,背景扫描为空气。采用溴化钾压片法制样,将浓度约为0.01mg
·
ml-1
的聚合物分散液滴在溴化钾上,充分干燥后测试。
[0068]
(5)热重分析
[0069]
测试仪器为美国ta公司生产的discovery tga55型热重分析仪。测试程序设置为:室温升温至100℃,升温速率为20℃/min,恒温10min;然后从100℃升温至780℃,升温速率为20℃/min。测试气氛为氮气,样品用量4
–
10mg。
[0070]
3、测试结果的比较与分析
[0071]
图2(a)给出了在实施例1中所合成的hbpe@acryl@poss的外观照片,常温下该样品呈固态,颜色为乳白色;作为对比,图2(b)同时给出了在比较例1中合成的hbpe@acryl的外观图片,该样品室温下呈半流动态,但几乎没有颜色,为半透明样品。上述外观的差异初步表明通过实施例1所述工艺已将poss基团引入hbpe结构中。图2(a)给出了所述hbpe@acryl(实施例1)的1hnmr谱图,证实已经将多个poss基团接枝入hbpe中,比例达4.39mol%(即每100个乙烯结构单元中含4.39个poss基团)。图2(a)和(b)的gpc流出曲线表明了两种聚合物分子量分布都比较窄。图3a给出了实施例1和比较例1制备的聚合物的红外分析图,在2925cm-1
和2854cm-1
处有明显的饱和碳氢单键的伸缩振动峰,对应两者超支化结构上甲基、亚甲基及次甲基基团的振动峰。同时hbpe@acryl@poss在1742cm-1
和1114cm-1
处分别对应碳氧双键的伸缩振动峰和硅氧单键的反对称伸缩振动峰,这两处振动峰是poss的特征峰,充分说明超支化聚合物接枝有poss单体。图3b给出了实施例1和比较例1制备的聚合物的x射线衍射分析图,比较例1合成的hbpe@acryl由于其独特的超支化结构,不具有结晶区域,呈现非晶油状液体状态,只有在2θ=18.7
°
只有一个较宽的衍射峰。而纯poss基团由于规整的硅氧六面体结构,在2θ=8.0
°
有很强的尖锐的衍射峰。图3c给出了实施例1和比较例1制备的聚合物的热重分析图,从图上可以知道两者的分解温度都在400℃左右。且在800℃条件下热分解完成,hbpe@acryl@poss经过热重所残留的si原子的质量分数占比为2.181%。
[0072]
实施例2、比较例2
[0073]
1、样品的制备
[0074]
(1)实施例2样品的制备按如下步骤进行:
[0075]
第1步:在100ml大小的柱状玻璃瓶内,依次加入500目天然鳞片状石墨(800mg)、分析纯氯仿(80ml)和由上述实施例1所得hbpe@acryl@poss(160mg),密封后置于250w超声水池中于25℃下恒温超声48h,获得石墨烯初始分散液,进一步在4000rpm下低速离心45min,静置2h后吸取上部2/3碳纳米管分散液,获得含过量hbpe@acryl@poss的石墨烯分散液(注:石墨粉的投料浓度为10mg/ml,hbpe@acryl@poss的投料浓度为2mg/ml)。用孔径为0.1μm的聚四氟乙烯抽滤膜进行真空抽滤除去过量的聚合物,随后将沉积石墨烯的抽滤膜取出,用新鲜的氯仿进行超声,此过程重复2次,得到除去过量聚合物的hbpe@acryl@poss基石墨烯分散液。
[0076]
第2步:在100ml大小的柱状玻璃瓶内,依次加入平均长度20nm的多壁碳纳米管(100mg)、分析纯氯仿(80ml)和由上述实施例1所得hbpe@acryl@poss(160mg),密封后置于250w超声水池中于25℃下恒温超声12h,获得碳纳米管初始分散液,进一步在4000rpm下低速离心45min,静置2h后吸取上部2/3碳纳米管分散液,,获得含过量hbpe@acryl@poss的碳纳米管分散液(注:碳纳米管的投料浓度为1.25mg/ml,hbpe@acryl@poss的投料浓度为2mg/ml)。用孔径为0.1μm的聚四氟乙烯抽滤膜进行真空抽滤除去过量的聚合物,随后将沉积碳纳米管的抽滤膜取出,用新鲜的氯仿进行超声,此过程重复2次,得到除去过量聚合物的hbpe@acryl@poss基碳纳米管分散液。
[0077]
(2)比较例2样品的制备按如下步骤进行:
[0078]
第1步:在100ml大小的柱状玻璃瓶内,依次加入500目天然鳞片状石墨(800mg)、分
析纯氯仿(80ml)和由上述比较例1所得hbpe@acryl(160mg),密封后置于250w超声水池中于25℃下恒温超声48h,获得石墨烯初始分散液,进一步在4000rpm下低速离心45min,静置2h后吸取上部2/3碳纳米管分散液,获得含过量hbpe@acryl的石墨烯分散液(注:石墨粉的投料浓度为10mg/ml,hbpe@acryl的投料浓度为2mg/ml)。对上述石墨烯分散液进行收集,用孔径为0.1μm的聚四氟乙烯抽滤膜进行真空抽滤除去过量的聚合物,随后将沉积石墨烯的抽滤膜取出,用新鲜的氯仿进行超声,此过程重复2次,得到除去过量聚合物的hbpe@acryl基石墨烯分散液。
[0079]
第2步:在100ml大小的柱状玻璃瓶内,依次加入平均长度20nm的多壁碳纳米管(100mg)、分析纯氯仿(80ml)和由上述比较例1所得hbpe@acryl(160mg),密封后置于250w超声水池中于25℃下恒温超声12h,获得碳纳米管初始分散液,进一步在4000rpm下低速离心45min,静置2h后吸取上部2/3碳纳米管分散液,获得含过量hbpe@acryl的碳纳米管分散液(注:碳纳米管的投料浓度为1.25mg/ml,hbpe@acryl的投料浓度为2mg/ml)。对上述碳纳米管分散液进行收集,用孔径为0.1μm的聚四氟乙烯抽滤膜进行真空抽滤除去过量的聚合物,随后将沉积碳纳米管的抽滤膜取出,用新鲜的氯仿进行超声,此过程重复2次,得到除去过量聚合物的hbpe@acryl基碳纳米管分散液。
[0080]
2、表征与测试
[0081]
(1)x射线衍射分析
[0082]
测试仪器为荷兰pnalytical公司生产的x pert pro型x射线衍射分析仪。x射线源为cu靶kα射线测试电压40kv,电流40ma,扫描速度10
°
/min,扫描范围5
–
80
°
。聚合物分散液滴在玻璃片上,溶剂充分干燥后直接用于测试。
[0083]
(2)热重分析
[0084]
测试仪器为美国ta公司生产的discovery tga55型热重分析仪。测试程序设置为:室温升温至100℃,升温速率为20℃/min,恒温10min;然后从100℃升温至780℃,升温速率为20℃/min。测试气氛为氮气,样品用量4
–
10mg。
[0085]
3、测试结果的比较与分析
[0086]
图4a给出了实施例2所得到的分散液中石墨烯、碳纳米管浓度分别为0.33mg/ml和0.39mg/ml;图4b给出了比较例2所得到的分散液中石墨烯、碳纳米管浓度分别为0.19mg/ml和0.32mg/ml。图4(c)表明实施例2和对比例2制备所得的石墨烯分散液均在2θ=26.30
°
附近出现了(002)晶面衍射峰,表明了石墨烯的成功制备。
[0087]
实施例3、比较例3
[0088]
1、样品的制备
[0089]
(1)实施例3样品的制备按如下步骤进行:
[0090]
第1步:制备碳材料总体积为3.05mm3的电热涂层(注:圆片状,直径43mm,厚度2.1μm,石墨烯与碳纳米管体积比为4:1):使用移液枪取实施例2制备的体积16.26ml(5.37mg)hbpe@acryl@poss基石墨烯分散液与2.84ml(1.10mg)hbpe@acryl@poss基碳纳米管分散液(注:热重分析已经得出石墨烯分散液的浓度为0.33mg/ml,碳纳米管分散液的浓度为0.39mg/ml;石墨烯和碳纳米管密度分别为2.2g/cm3和1.8g/cm3)并添加3mg/ml 2,2-二甲氧基-2-苯基苯乙酮,超声5min混合均匀后,抽滤在ptfe(孔径100nm)膜上,然后待膜层中的溶剂自然挥发后得到固态膜。
[0091]
第2步:利用1kw手提式uv固化机对涂层进行紫外光固化5min得到一张致密的电热涂层。(具体操作如图5所示)
[0092]
(2)比较例3样品的制备按如下步骤进行:
[0093]
第1步:制备碳材料总体积为3.05mm3(注:圆片状,直径43mm,厚度2.1μm,石墨烯与碳纳米管体积比为4:1)的电热涂层:使用移液枪取比较例2制备的体积27.9ml(5.30mg)hbpe@acryl基石墨烯分散液与3.41ml(1.09mg)hbpe@acryl基碳纳米管分散液(热重分析已经得出石墨烯分散液浓度为0.19mg/ml,碳纳米管分散液的浓度为0.32mg/ml;石墨烯和碳纳米管密度分别为2.2g/cm3和1.8g/cm3)并添加3mg/ml 2,2-二甲氧基-2-苯基苯乙酮,超声5min混合均匀后,抽滤在ptfe(孔径100nm)膜上,然后待膜层中的溶剂自然挥发后得到固态膜。
[0094]
第2步:利用1kw手提式uv固化机对涂层进行紫外光固化5min得到一张致密的电热涂层。
[0095]
2、表征与测试
[0096]
(1)四探针测试:测试仪器为广州四探针科技有限公司生产的rts-8型四探针测试仪。测试条件:根据方阻(方块电阻)大小选择电流量程,其中方阻大于100kω选择1μa,测试电流为0.4459μa;1kω~100kω,选择10μa,测试电流为4.459μa;小于1kω选择100μa,测试电流为44.59μa。探针间距为1mm。
[0097]
测试方法:将样品放置在测试台上,对2个样品表面目标区域进行测试,每个样品测试10次,取平均值。
[0098]
(2)红外热成像测试:采用美国fluke公司生产的ti55ft-20型红外热像仪首先将制得的导电膜裁成一个长方形(2
×
3.4cm2),将铜片(2
×
1.1cm2)垫在样品短边,鳄鱼夹夹住后,通直流电并调节不同的电压,并用红外热成像仪进行观察。
[0099]
(3)电热涂层微观形貌分析:采用美国fei公司生产的nova nano sem 450型场发射扫描电子显微镜对样品表面进行观察。样品的制备:将所观察区域剪出,粘在导电胶上,再将导电胶粘在表面样品台上,再做喷金处理得到测试样品。
[0100]
(4)比热测试:将涂层从ptfe膜片上刮取下来,在n2保护条件下以10℃/min进行升温,从0℃升温至60℃进行测试。
[0101]
3、测试结果的比较与分析
[0102]
图6(a)给出了实施例3和比较例3制备的电热涂层在相同构成情况下方块电阻的测试结果,相对于比较例3,引入poss基团的实施例3具有更低的方块电阻;图6(b)给出了不同电压下实施例3的hbpe@acryl@poss基电热涂层和比较例3的hbpe@acryl基电热涂层的温度变化情况,相对于hbpe@acryl基电热涂层,hbpe@acryl@poss基电热涂层具有更高的电热温度和耐温性能(180℃条件下涂层温度保持稳定,hbpe@acryl基电热涂层只能在140℃条件下涂层温度保持稳定);图6(c)表明了实施例3与比较例3的电热涂层的极限耐温电压图像,相对于比较例3的hbpe@acryl基电热涂层,实施例3的hbpe@acryl@poss基电热涂层具有更高的电压承载能力。图6(d)表明了实施例3表面的poss基团的分布非常的均匀,这也保证了基团功能性显示。
[0103]
图7(a)与(b)表明在不同电压情况下实施例3的hbpe@acryl@poss基电热涂层与比较例3的hbpe@acryl基电热涂层升温速率与降温速率的不同。实施例3的升温速率和降温速
率都大于比较例3的升温速率与降温速率。图7(c)则是实施例3与比较例3的电热涂层的动态比热图,可以从动态比热图中看出,poss基团的引入可以降低碳基涂层的比热,根据公式1可知比热越大升温、降温速率越慢,所以引入poss基团对于降低碳基电热涂层比热增大升温降温速率具有正面意义。
[0104]
公式1:
[0105]
实施例4、比较例4
[0106]
1、样品的制备
[0107]
(1)实施例4样品的制备按如下步骤进行:
[0108]
第1步:将实施例3的hbpe@acryl@poss基电热涂层裁成长方形(2
×
3.4cm2),对样品进行弯折处理,具体如图8所示。其中,先面内弯折一次然后面外弯折一次,记录为交替弯折一次。
[0109]
第2步:每进行300次弯折进行一次涂层电热性能测试。
[0110]
(2)比较例4样品的制备按如下步骤进行:
[0111]
第1步:将比较例3的hbpe@acryl基电热涂层裁成长方形(2
×
3.4cm2),对样品进行弯折处理。其中,先面内弯折一次然后面外弯折一次,记录为交替弯折一次。
[0112]
第2步:每进行300次弯折进行一次涂层电热性能测试。
[0113]
2、表征与测试
[0114]
(1)红外热成像测试:采用首先将制得的导电膜裁成一个长方形(2
×
3.4cm2),将铜片(2
×
1.1cm2)垫在样品短边,鳄鱼夹夹住后,通直流电并调节不同的电压,并用红外热成像仪进行观察。
[0115]
(2)电热涂层微观形貌分析:采用美国fei公司生产的nova nano sem 450型场发射扫描电子显微镜对样品表面进行观察。样品的制备:将所观察弯折区域剪出,粘在导电胶上,再将导电胶粘在表面样品台上,再做喷金处理得到测试样品。
[0116]
3、测试结果的比较与分析
[0117]
图9(a)是实施例4为hbpe@acryl@poss基电热涂层和比较例4的hbpe@acryl基电热涂层弯折前后的数码照片。图9(b)为在18v条件下实施例4的hbpe@acryl@poss基电热涂层和比较例4的hbpe@acryl基电热涂层在不同弯折次数下电热性能的表现情况,从9(b)可知,实施例4的hbpe@acryl@poss基电热涂层在弯折2700次条件下依旧保持电热温度的稳定,而比较例4的hbpe@acryl基电热涂层则在弯折2100次条件下完全丧失电热性能。实施例4的hbpe@acryl@poss基电热涂层具有更好的耐弯折性能。
[0118]
实施例5、比较例5
[0119]
1、样品的制备
[0120]
(1)实施例5样品的制备按如下步骤进行:
[0121]
第1步:在乙烯气体的保护下,在250ml的schlenk瓶中依次加入6g poss单体(0.32mol/l)和0.32g 1,4-丁二醇二丙烯酸酯单体(0.08mol/l),随后加入pd-diimine催化剂250mg溶解于10ml无水二氯甲烷当中,利用油浴锅控制反应的温度在30℃,随后加入10ml无水二氯甲烷在600r/min的搅拌下反应24h。聚合反应24h后,将所得到的产物转移到200ml的酸化甲醇当中,用于终止聚合反应。
[0122]
第2步:将所得的聚合产物进行纯化,具体操作如下:在常温条件下吹除溶剂,随后分别滴加20滴双氧水和稀盐酸并加入50ml四氢呋喃搅拌2h去除聚合物中的pd颗粒;随后加入100ml甲醇使聚合产物沉淀析出。随后再加入20ml四氢呋喃对产物进行溶解,再用甲醇重新沉淀,该步骤重复2~3次;加入20ml甲苯对产物进行溶解,再用甲醇重新沉淀,该步骤重复2~3次,所得产物经30℃下真空干燥48h后获得hbpe@acryl@poss。
[0123]
(2)比较例5样品的制备按如下步骤进行:
[0124]
在乙烯气体的保护下,在250ml的schlenk瓶中依次加入20g poss单体(0.864mol/l)和3g 1,4-丁二醇二丙烯酸酯单体(0.576mol/l),随后加入pd-diimine催化剂200mg溶解于10ml无水二氯甲烷当中,利用油浴锅控制反应的温度在30℃,随后加入10ml无水二氯甲烷在600r/min的搅拌下反应24h。聚合反应24h后,将所得到的产物转移到200ml的酸化甲醇当中,用于终止聚合反应。
[0125]
第2步:将所得的聚合产物进行纯化,具体操作如下:在常温条件下吹除溶剂,随后分别滴加20滴双氧水和稀盐酸并加入50ml四氢呋喃搅拌2h去除聚合物中的pd颗粒;随后加入100ml甲醇使聚合产物沉淀析出。随后再加入20ml四氢呋喃对产物进行溶解,再用甲醇重新沉淀,该步骤重复2~3次;加入20ml甲苯对产物进行溶解,再用甲醇重新沉淀,该步骤重复2~3次,所得产物经30℃下真空干燥48h后获得hbpe@acryl@poss。
[0126]
2、表征与测试
[0127]
(1)1h核磁共振波谱测试
[0128]
实施例5与比较例5的hbpe@acryl@poss 1
h核磁共振波谱(1h nmr)由500mhz anance iii型核磁共振仪(瑞士bruker公司)测得,溶剂为氘代氯仿,测试温度为室温。
[0129]
(2)凝胶渗透色谱分析
[0130]
测试仪器为英国malvern公司生产的omnisec型凝胶渗透色谱仪。测试温度为30℃,ps为标样,thf为流动相。样品制备:聚合物配置成浓度为5mg
·
ml-1
的thf溶液,体积6ml左右,用孔径为0.45微米的有机膜过滤后用于测试。
[0131]
3、测试结果的比较与分析
[0132]
图10(a)给出了在实施例5中所合成的hbpe@acryl@poss的外观照片,作为对比,图10(b)同时给出了在比较例5中合成的hbpe@acryl@poss的外观图片,两者在常温下都呈固态,颜色为乳白色,无明显差别。图10(a)给出了hbpe@acryl@poss(实施例5)的1hnmr谱图,证明更改投料比可以更改丙烯酰氧基和poss基的接枝率(实施例5中1,4-丁二醇二丙烯酸酯与poss的投料比为1:4,比较例5中1,4-丁二醇二丙烯酸酯与poss的投料比为1:1.5),实施例5中的poss接枝率达8.20mol%(即每100个乙烯结构单元中含8.20个poss基团)。图10(a)和(b)的gpc流出曲线表明了poss接枝率的提高会使聚合物分子量分布变宽。
[0133]
实施例6、比较例6
[0134]
1、样品的制备
[0135]
(1)实施例6样品的制备按如下步骤进行:
[0136]
第1步:在100ml大小的柱状玻璃瓶内,依次加入500目天然鳞片状石墨(800mg)、分析纯氯仿(80ml)和由上述实施例5所得hbpe@acryl@poss(160mg),密封后置于250w超声水池中于25℃下恒温超声48h,获得石墨烯初始分散液,进一步在4000rpm下低速离心45min,随后静置2h,获得含过量hbpe@acryl@poss的石墨烯分散液(注:石墨粉的投料浓度为10mg/
ml,hbpe@acryl@poss的投料浓度为2mg/ml)。用孔径为0.1μm的聚四氟乙烯抽滤膜进行真空抽滤除去过量的聚合物,随后将沉积石墨烯的抽滤膜取出,用新鲜的氯仿进行超声,此过程重复2次,得到除去过量聚合物的hbpe@acryl@poss基石墨烯分散液。
[0137]
第2步:在100ml大小的柱状玻璃瓶内,依次加入平均长度20nm的多壁碳纳米管(100mg)、分析纯氯仿(80ml)和由上述实施例5所得hbpe@acryl@poss(160mg),密封后置于250w超声水池中于25℃下恒温超声12h,获得碳纳米管初始分散液,进一步在4000rpm下低速离心45min,随后静置2h,获得含过量hbpe@acryl@poss的碳纳米管分散液(注:碳纳米管的投料浓度为1.25mg/ml,hbpe@acryl@poss的投料浓度为2mg/ml)。用孔径为0.1μm的聚四氟乙烯抽滤膜进行真空抽滤除去过量的聚合物,随后将沉积碳纳米管的抽滤膜取出,用新鲜的氯仿进行超声,此过程重复2次,得到除去过量聚合物的hbpe@acryl@poss基碳纳米管分散液。
[0138]
(2)比较例6样品的制备按如下步骤进行:
[0139]
第1步:在100ml大小的柱状玻璃瓶内,依次加入500目天然鳞片状石墨(800mg)、分析纯氯仿(80ml)和由上述比较例5所得hbpe@acryl@poss(160mg),密封后置于250w超声水池中于25℃下恒温超声48h,获得石墨烯初始分散液,进一步在4000rpm下低速离心45min,随后静置2h,获得含过量hbpe@acryl@poss的石墨烯分散液(注:石墨粉的投料浓度为10mg/ml,hbpe@acryl@poss的投料浓度为2mg/ml)。用孔径为0.1μm的聚四氟乙烯抽滤膜进行真空抽滤除去过量的聚合物,随后将沉积石墨烯的抽滤膜取出,用新鲜的氯仿进行超声,此过程重复2次,得到除去过量聚合物的hbpe@acryl@poss基石墨烯分散液。
[0140]
第2步:在100ml大小的柱状玻璃瓶内,依次加入平均长度20nm的多壁碳纳米管(100mg)、分析纯氯仿(80ml)和由上述比较例5所得hbpe@acryl@poss(160mg),密封后置于250w超声水池中于25℃下恒温超声12h,获得碳纳米管初始分散液,进一步在4000rpm下低速离心45min,随后静置2h,获得含过量hbpe@acryl@poss的碳纳米管分散液(注:碳纳米管的投料浓度为1.25mg/ml,hbpe@acryl@poss的投料浓度为2mg/ml)。用孔径为0.1μm的聚四氟乙烯抽滤膜进行真空抽滤除去过量的聚合物,随后将沉积碳纳米管的抽滤膜取出,用新鲜的氯仿进行超声,此过程重复2次,得到除去过量聚合物的hbpe@acryl@poss基碳纳米管分散液。
[0141]
2、表征与测试
[0142]
(1)热重分析
[0143]
测试仪器为美国ta公司生产的discovery tga55型热重分析仪。测试程序设置为:室温升温至100℃,升温速率为20℃/min,恒温10min;然后从100℃升温至780℃,升温速率为20℃/min。测试气氛为氮气,样品用量4~10mg。
[0144]
3、测试结果的比较与分析
[0145]
图11(a)给出了实施例6所得到的石墨烯、碳纳米管浓度分别为0.66mg/ml和0.46mg/ml;图11(b)给出了比较例6所得到的石墨烯、碳纳米管浓度分别为0.33mg/ml和0.39mg/ml。
[0146]
实施例7、比较例7
[0147]
1、样品的制备
[0148]
(1)实施例7样品的制备按如下步骤进行:
[0149]
第1步:制备碳材料总体积为3.05mm3(注:圆片状,直径43mm,厚度2.1μm,石墨烯与碳纳米管体积比为4:1)的电热涂层,使用移液枪取实施例6制备的体积8.1ml(5.35mg)hbpe@acryl@poss基石墨烯分散液与2.4ml(1.10mg)hbpe@acryl@poss基碳纳米管分散液(注:热重分析已经得出石墨烯分散液的浓度为0.66mg/ml,碳纳米管分散液的浓度为0.46mg/ml;石墨烯和碳纳米管密度分别为2.2g/cm3和1.8g/cm3)并添加3mg/ml 2,2-二甲氧基-2-苯基苯乙酮,超声5min混合均匀后,抽滤在ptfe(孔径100nm)膜上,然后待膜层中的溶剂自然挥发后得到固态膜。
[0150]
第2步:利用手提式uv固化机对涂层进行紫外光固化5min得到一张致密的电热涂层。
[0151]
(具体操作如图5所示)
[0152]
(2)比较例7样品的制备按如下步骤进行:
[0153]
第1步:制备碳材料总体积为3.05mm3(注:直径43mm,厚度2.1μm,石墨烯与碳纳米管体积比为4:1)的电热涂层,使用移液枪取比较例6制备的体积16.26ml(5.37mg)hbpe@acryl@poss基石墨烯分散液与2.84ml(1.10mg)hbpe@acryl@poss基碳纳米管分散液(注:热重分析已经得出石墨烯分散液的浓度为0.33mg/ml,碳纳米管分散液的浓度为0.39mg/ml;石墨烯和碳纳米管密度分别为2.2g/cm3和1.8g/cm3)并添加3mg/ml 2,2-二甲氧基-2-苯基苯乙酮,超声5min混合均匀后,抽滤在ptfe(孔径100nm)膜上,然后待膜层中的溶剂自然挥发后得到固态膜。
[0154]
第2步:利用手提式uv固化机对涂层进行紫外光固化5min得到一张致密的电热涂层。
[0155]
(具体操作如图5所示)
[0156]
2、表征与测试
[0157]
(1)四探针测试:测试仪器为广州四探针科技有限公司生产的rts-8型四探针测试仪。测试条件:根据方阻(方块电阻)大小选择电流量程,其中方阻大于100kω选择1μa,测试电流为0.4459μa;1kω~100kω,选择10μa,测试电流为4.459μa;小于1kω选择100μa,测试电流为44.59μa。探针间距为1mm。
[0158]
测试方法:将样品放置在测试台上,对2个样品表面目标区域进行测试,每个样品测试10次,取平均值。
[0159]
(2)红外热成像测试:采用美国fluke公司生产的ti55ft-20型红外热像仪首先将制得的导电膜裁成一个长方形(2
×
3.4cm2),将铜片(2
×
1.1cm2)垫在样品短边,鳄鱼夹夹住后,通直流电并调节不同的电压,并用红外热成像仪进行观察。
[0160]
3、测试结果的比较与分析
[0161]
图12(a)给出了实施例7和比较例7在相同构成情况下方块电阻的测试结果,相对于比较例7,实施例7中poss基团接枝率的提高对方块电阻基本不产生变化;图12(b)给出了不同电压下实施例7和比较例7的hbpe@acryl@poss基电热涂层的温度变化情况,相对于低poss接枝率hbpe@acryl@poss基电热涂层,高接枝率hbpe@acryl@poss基电热涂层具有更高的电热温度和耐温性能(280℃条件下涂层温度保持稳定,低接枝率hbpe@acryl@poss基电热涂层只能在180℃条件下涂层温度保持稳定);图12(c)表明了实施例7与比较例7的极限耐温电压图像,相对于比较例7的低接枝率hbpe@acryl@poss基电热涂层,实施例7的高接枝
率hbpe@acryl@poss基电热涂层具有更高的电压承载能力,能够达到29v。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1