一种生物基可降解嵌段共聚酯的制备方法及其在压敏胶方面的应用
1.本发明涉及高分子材料以及化学化工领域,具体地,涉及一种聚乳酸-b-聚(δ位取代戊内酯)-b-聚乳酸嵌段共聚物的制备方法及其在压敏胶方面的应用。
背景技术:
2.压敏胶是兼具固体弹性和液体粘性的一类高分子材料,它可以通过轻轻按压就能够粘附在物体表面且不与被粘物体发生化学反应。压敏胶操作简单,能够与被黏物形成牢固的非永久结合,被广泛应用于包装、汽车、医疗、电子等多个领域。然而现在所使用的压敏胶大多是丙烯酸酯类或苯乙烯嵌段共聚物,废弃后在自然条件下难以降解,会造成环境污染。因此研发可在自然环境下降解的压敏胶十分必要。
3.δ位取代戊内酯天然存在于牛乳、椰子中,具有芳香气味,通常被用作香料和食品添加剂。l-丙交酯可以由富含糖类的生物质发酵得到,由其开环聚合得到的聚乳酸是一种生物可降解聚酯材料。通过选择合适的催化体系,使用一锅法顺序开环聚合δ位取代戊内酯和l-丙交酯可以制备得到结构明确的聚乳酸-b-聚(δ位取代戊内酯)-b-聚乳酸三嵌段共聚物。通过改变δ位取代基的长度,可以调控所得三嵌段共聚物的缠结摩尔质量、玻璃化转变温度和粘弹性质,有望在压敏胶方面取得应用。
4.本发明提供了一种聚乳酸-b-聚(δ位取代戊内酯)-b-聚乳酸嵌段共聚物的制备方法及其在压敏胶方面的应用。本发明提供的方法与以往报道的方法相比,具有以下有益之处:1)制备得到的嵌段共聚物可以直接作为压敏胶使用,无需额外添加增粘剂、增塑剂等其他助剂,在自然条件下可完全降解;2)原料来源于生物质;3)制备得到的压敏胶适用于各类纸质基材和塑料薄膜类基材,具有和商品化压敏胶带近似甚至更好的剥离强度。
技术实现要素:
5.本发明的目的是提供一种生物基可降解嵌段共聚酯的制备方法及其在压敏胶方面的应用,以解决现有压敏胶在自然条件下不能完全降解的问题。
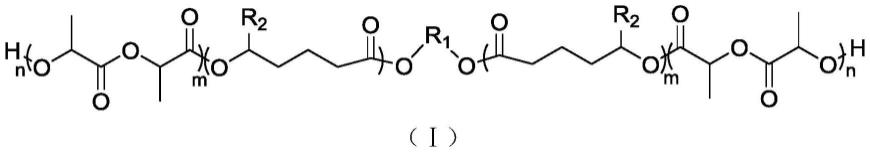
6.本发明提供式(ⅰ)所示聚乳酸-b-聚(δ位取代戊内酯)-b-聚乳酸三嵌段共聚物,其中r1为式(ⅱ)、式(ⅲ)、式(ⅳ)、式(
ⅴ
)、式(ⅵ)、式(ⅶ)、式(
ⅷ
)、式(
ⅸ
)所示亚烷基或芳基亚烷基中的一种;r2为正丙基、正己基、正壬基中的一种;m为50~150的自然数,n为10~30的自然数。
7.[0008][0009]
本发明还提供了上述聚乳酸-b-聚(δ位取代戊内酯)-b-聚乳酸三嵌段共聚物的制备方法,包括如下步骤:
[0010]
(1)将引发剂、强碱和助催化剂溶于有机溶剂中,在室温下搅拌1~5min;
[0011]
(2)将δ位取代戊内酯加入上述混合溶液中,在0~60℃下反应1~8h;将丙交酯加入上述反应体系,在0~60℃下继续反应0.1~1h,加入酸性物质终止反应,将反应混合物加入甲醇中沉淀得到聚乳酸-b-聚(δ位取代戊内酯)-b-聚乳酸三嵌段共聚物。
[0012]
上述制备方法中,所述助催化剂具有式(x)所示结构:
[0013][0014]
上述的制备方法中,所述引发剂为二元醇,具体可为乙二醇、1,2-丙二醇、1,2-丁二醇、1,4-丁二醇、2-丁基-2-乙基-1,3-丙二醇、1,2-苯二甲醇、1,3-苯二甲醇、1,4-苯二甲醇;所述强碱可为碱金属、碱金属化合物或有机磷腈碱催化剂,具体可为钠、钾、氢化钾、氢化钠、六[三(二甲基胺)磷氮烯]三聚磷腈({[(nme2)3p=n]2p=n}3)、磷腈配体p4-叔丁基([(nme2)3p=n]3p=ntbu,tert-bu-p4)、磷腈配体p2-叔丁基([(nme2)3p=n](nme2)2p=ntbu,tert-bu-p2);所述δ位取代戊内酯为δ-辛内酯、δ-十一内酯、δ-十四内酯;所述有机溶剂为甲苯、四氢呋喃、二氯甲烷、三氯甲烷、二氧六环、乙腈、n,n-二甲基甲酰胺;所述酸性物质为乙酸、苯甲酸、盐酸、硫酸、磷酸中的任意一种。
[0015]
所述强碱与引发剂的摩尔比例为1/1~10/1;所述强碱与助催化剂的摩尔比例为1/1~1/5;所述δ位取代戊内酯与引发剂的摩尔比例为100/1~300/1;所述丙交酯与引发剂的摩尔比例为20/1~60/1;所述酸性物质与催化剂的摩尔比例为1/1~10/1。
[0016]
所述δ位取代戊内酯的摩尔浓度为1~9mol/l;所述丙交酯的摩尔浓度为0.1~2mol/l。
[0017]
本发明的另一目的是提供基于上述聚乳酸-b-聚(δ位取代戊内酯)-b-聚乳酸三嵌段共聚物的压敏胶带的制备方法,包括如下步骤:
[0018]
将聚乳酸-b-聚(δ位取代戊内酯)-b-聚乳酸三嵌段共聚物涂布在基底薄膜上,并在25℃,50%湿度的恒温恒湿箱中调节24h,得到压敏胶带;所述基底薄膜为涤纶树脂薄膜(pet)、聚酰亚胺薄膜(pi)、聚氯乙烯薄膜(pvc)、双向拉伸聚丙烯薄膜(bopp)中的任意一
种。
附图说明
[0019]
图1为对比实施例1制备聚乳酸-b-聚(δ-己内酯)-b-聚乳酸嵌段共聚物的1h nmr谱图。
[0020]
图2为实施例1-3制备的聚乳酸-b-聚(δ位取代戊内酯)-b-聚乳酸嵌段共聚物gpc谱图。
[0021]
图3为实施例1制备的聚乳酸-b-聚(δ-辛内酯)-b-聚乳酸嵌段共聚物的1h nmr谱图。
[0022]
图4为实施例1制备的聚乳酸-b-聚(δ-辛内酯)-b-聚乳酸嵌段共聚物的
13
c nmr谱图。
[0023]
图5为实施例2制备的聚乳酸-b-聚(δ-十一内酯)-b-聚乳酸嵌段共聚物的1h nmr谱图。
[0024]
图6为实施例2制备的聚乳酸-b-聚(δ-十一内酯)-b-聚乳酸嵌段共聚物的
13
c nmr谱图。
[0025]
图7为实施例3制备的聚乳酸-b-聚(δ-十四内酯)-b-聚乳酸嵌段共聚物的1h nmr谱图。
[0026]
图8为实施例3制备的聚乳酸-b-聚(δ-十四内酯)-b-聚乳酸嵌段共聚物的
13
c nmr谱图。
具体实施方式
[0027]
下述实施案例对本发明进行具体描述,但本发明不限于这些实施案例。
[0028]
下述实施案例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0029]
对比实施例1
[0030]
将(0.08mmol,11.05mg)1,2-苯二甲醇,(0.08mmol,95.84mg)六[三(二甲基胺)磷氮烯]三聚磷腈,(0.08mmol,47.82mg)1,1'-(氧双(亚乙基))双(3-(3,5-二(三氟甲基)苯基)脲溶于2.8ml四氢呋喃中,在室温下搅拌10min,将(16mmol,1.8ml)δ-己内酯加入反应管中,反应在30℃氮气保护下进行120min。将(4mmol,0.56g)丙交酯溶于10.7ml四氢呋喃,加入上述体系,在30℃下氮气保护反应30min,加入10mg苯甲酸终止反应。将反应混合物倒入60ml甲醇中,离心分离沉淀得到聚合物,聚合物1h nmr谱图如图1所示。制备得到的三嵌段共聚物表现为塑料性质,无法润湿被粘接表面,无法直接作为压敏胶使用。
[0031]
对比实施例2
[0032]
将(0.08mmol,11.05mg)1,2-苯二甲醇,(0.08mmol,95.84mg)六[三(二甲基胺)磷氮烯]三聚磷腈,(0.08mmol,38.7mg)1,3-(3,5-双三氟甲基苯基)脲溶于2.8ml四氢呋喃中,在室温下搅拌10min,将(16mmol,2.4ml)δ-辛内酯加入反应管中,反应在30℃氮气保护下进行120min。将(4mmol,0.56g)丙交酯溶于10.7ml四氢呋喃,加入上述体
系,在30℃下氮气保护反应30min,加入10mg苯甲酸终止反应。将反应混合物倒入60ml甲醇中,没有得到聚合物。核磁氢谱表明δ-辛内酯和丙交酯的转化率均低于2%。
[0033]
实施例1
[0034]
将(0.08mmol,11.05mg)1,2-苯二甲醇,(0.08mmol,95.84mg)六[三(二甲基胺)磷氮烯]三聚磷腈,(0.08mmol,47.82mg)1,1'-(氧双(亚乙基))双(3-(3,5-二(三氟甲基)苯基)脲溶于2.8ml四氢呋喃中,在室温下搅拌10min,将(16mmol,2.4ml)δ-辛内酯加入反应管中,反应在30℃氮气保护下进行120min。将(4mmol,0.56g)丙交酯溶于10.7ml四氢呋喃,加入上述体系,在30℃下氮气保护反应30min,加入10mg苯甲酸终止反应。将反应混合物倒入60ml甲醇中,离心分离沉淀得到聚合物,gpc测得数均分子量为30.2kg/mol,分子量分布为1.16,gpc谱图如图2所示,1h nmr谱图如图3所示,
13
c nmr谱图如图4所示。所得三嵌段共聚物在室温下具有一定的流动性,利用涂布器将其涂布在pet薄膜上,并在25℃,50%湿度的恒温恒湿箱中调节24h,得到压敏胶带,按照gb/t2792方法测试胶带的180
°
剥离强度。
[0035]
实施例2
[0036]
将(0.16mmol,22.10mg)1,2-苯二甲醇,(0.16mmol,58.8mg)磷腈配体p2-叔丁基催化剂,(0.16mmol,95.64mg)1,1'-(氧双(亚乙基))双(3-(3,5-二(三氟甲基)苯基)脲溶于5.6ml四氢呋喃中,在室温下搅拌10min,将(32mmol,6.2ml)δ-十一内酯加入反应管中,反应在30℃氮气保护下进行120min。将(8mmol,1.12g)丙交酯溶于21.4ml四氢呋喃,加入上述体系,在30℃下氮气保护反应30min,加入1ml醋酸终止反应。将反应混合物倒入150ml甲醇中,离心分离沉淀得到聚合物,gpc测得数均分子量为32.4kg/mol,分子量分布为1.19,gpc谱图如图2所示。1h nmr谱图如图5所示,
13
c nmr谱图如图6所示。所得三嵌段共聚物在室温下具有一定的流动性,利用涂布器将其涂布在bopp薄膜上,并在25℃,50%湿度的恒温恒湿箱中调节24h,得到压敏胶带,按照gb/t2792测试胶带的180
°
剥离强度。
[0037]
实施例3
[0038]
将(0.08mmol,11.05mg)1,2-苯二甲醇,(0.08mmol,29.4mg)磷腈配体p2-叔丁基,(0.08mmol,47.82mg)1,1'-(氧双(亚乙基))双(3-(3,5-二(三氟甲基)苯基)脲溶于2.8ml四氢呋喃中,在室温下搅拌10min,将(16mmol,3.1ml)δ-十四内酯加入反应管中,反应在30℃氮气保护下进行120min。将(4mmol,0.56g)丙交酯溶于10.7ml四氢呋喃,加入上述体系,在30℃下氮气保护反应30min,加入1ml稀硫酸终止反应。将反应混合物倒入60ml甲醇中,离心分离沉淀得到聚合物,gpc测得数均分子量为37.8kg/mol,分子量分布为1.21,gpc谱图如图2所示。1h nmr谱图如图7所示,
13
c nmr谱图如图8所示。所得三嵌段共聚物在室温下具有一定的流动性,利用涂布器将其涂布在pvc薄膜上,并在25℃,50%湿度的恒温恒湿箱中调节24h,得到压敏胶带,按照gb/t2792测试胶带的180
°
剥离强度。
[0039]
实施例1-3制作的压敏胶带性能测试结果如表1所示:
[0040]
表1
[0041][0042]
由对比实施例1可以看出,δ位取代戊内酯的选择至关重要,并非所有的δ位取代戊内酯和丙交酯共聚得到的三嵌段共聚物都可以用作压敏胶。由对比实施例2可以看出,助催化剂的选择也很重要,不合适的助催化剂无法实现δ位取代戊内酯和丙交酯的顺序开环聚合,无法制备得到三嵌段共聚物。
[0043]
以上实施例仅是本发明的优选案例,并不对本发明造成限制,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1