作为CDK选择性抑制剂的新型哒嗪酮化合物的制作方法
作为cdk选择性抑制剂的新型哒嗪酮化合物
技术领域
1.本发明涉及可用作cdk选择性抑制剂的新型哒嗪酮化合物或其药学上可接受的盐。本发明还涉及包含一种或更多种此类化合物或其药学上可接受的盐作为活性成分的药物组合物,以及此类化合物或其药学上可接受的盐在治疗过度增殖性疾病例如癌症中的用途。
背景技术:
2.细胞周期蛋白依赖性激酶(cyclin dependent kinase,cdk)属于丝/苏氨酸蛋白激酶家族,是一种由细胞周期催化激酶亚基和调节亚基组成的异二聚体复合物,是参与细胞周期调节的关键激酶,其活性依赖于细胞周期蛋白的结合和激活,在调节细胞周期和基因转录中起关键作用(malumbres,m.(2014),“cyclin-dependent kinases.
”ꢀ
genome biol 15(6):122)。最初因其在调节细胞周期中的作用而被发现。根据cdk功能的不同,可以将其主要分为两大类。一类cdk参与细胞周期调控,主要包括cdk1、cdk2、cdk4、cdk6等;另一大类cdk参与转录调节,主要包括cdk7、cdk8、cdk9、cdk10、cdk11等。
3.肿瘤通常被认为是由一群增殖异常活跃的细胞所构成,其基本特征为过度活化、持续的细胞增殖,因此通过诱导细胞周期阻滞可有效抑制肿瘤的生长。
4.由于cdk活性为细胞分裂所必需,而在肿瘤细胞中又常有cdk活性增强,因此长期以来,cdk一直被认为是抗肿瘤及其他增殖失调疾病药物研发的较好靶点。cdk抑制剂对细胞周期控制起着至关重要的作用,可以阻断细胞的周期,控制细胞的增殖,从而达到抗肿瘤的目的。cdk靶向小分子抑制剂类药物具有很高的开发价值,发展空间大,对于在该领域内探索新的抗肿瘤药物具有重大的意义。
5.cdk抑制剂也可用于治疗心血管病症如再狭窄和动脉粥样硬化和由异常细胞增殖所致的其他血管病症。
6.目前已有的cdk抑制剂专利申请包括,如wo2015101293a1、wo2016015605a1、wo2016194831a1、wo2019161224a1、wo2020224568a1等等。由于巨大的市场需求,所以仍有必要继续研发低毒高效的cdk抑制剂,尤其是针对特定cdk具有较高选择性的cdk选择性抑制剂。
技术实现要素:
7.本发明旨在提供一类结构新颖(哒嗪酮化合物)的选择性地作用于cdk2/4的抑制剂。
8.在一方面,本发明涉及一种式(i)的化合物:
或其药学上可接受的盐,其中:环a是任选取代的含有0、1或2个选自o或n的杂原子的5-或6-元单环;环a与环b相稠合形成稠环;r1选自氢或c
1-6
烷基;r2选自氢或卤素;r3选自氢或氧代(=o);r4选自氢、c
1-6
烷基或-nrarb,其中ra和rb各自独立地选自氢或c
1-6
烷基;当代表双键时,x1选自c;当代表单键时,x1选自ch或n;x2、x3和x4各自独立地选自ch或n。
9.根据一些实施方案,在本发明的化合物或其药学上可接受的盐中,环a与环b相稠合形成选自以下的稠环:或,其余变量如本发明所定义。
10.根据一些实施方案,在本发明的化合物或其药学上可接受的盐中,结构单元选自或,其余变量如本发明所定义。
11.根据一些实施方案,本发明的化合物具有式(ia)所示的结构:
,其余变量如本发明所定义。
12.根据一些实施方案,本发明的化合物具有式(ib)所示的结构:,其余变量如本发明所定义。
13.根据一些实施方案,在本发明的化合物或其药学上可接受的盐中,r1选自氢或c
1-3
烷基,其余变量如本发明所定义。
14.根据一些实施方案,在本发明的化合物或其药学上可接受的盐中,r1选自氢、或,其余变量如本发明所定义。
15.根据一些实施方案,在本发明的化合物或其药学上可接受的盐中,r2选自氢、f、cl、br或i,其余变量如本发明所定义。
16.根据一些实施方案,在本发明的化合物或其药学上可接受的盐中,r3选自氢或氧代(=o),其余变量如本发明所定义。
17.根据一些实施方案,在本发明的化合物或其药学上可接受的盐中,r4选自氢、c
1-3
烷基或-nrarb,其中ra和rb各自独立地选自氢或c
1-3
烷基,其余变量如本发明所定义。
18.根据一些实施方案,在本发明的化合物或其药学上可接受的盐中,r4选自氢、甲基
或,其余变量如本发明所定义。
19.根据一些实施方案,在本发明的化合物或其药学上可接受的盐中,r1选自氢或c
1-3
烷基;r2选自氢、f、cl、br或i;r3选自氢或氧代(=o);r4选自氢、c
1-3
烷基或-nrarb,其中ra和rb各自独立地选自氢或c
1-3
烷基,其余变量如本发明所定义。
20.根据一些实施方案,在本发明的化合物或其药学上可接受的盐中,结构单元选自或,其余变量如本发明所定义。
21.根据一些实施方案,在本发明的化合物或其药学上可接受的盐中,结构单元选自、,或,其余变量如本发明所定义。
22.根据一些实施方案,在本发明的化合物或其药学上可接受的盐中,结构单元选自、、,或,其余变量如本发明所定义。
23.根据一些实施方案,本发明的化合物选自以下结构之一:
、、、,和。
24.在另一方面,本发明涉及一种药物组合物,该药物组合物包括如本文提供的式(i)的化合物或其药学上可接受的盐,和药学上可接受的载体或辅料。
25.在还一方面,本发明涉及一种治疗有需要的受试者的过度增殖性疾病例如癌症的方法,该方法包括向受试者施用治疗有效量的如本文提供的式(i)的化合物或其药学上可接受的盐。
26.在还一方面,本发明涉及如本文提供的式(i)的化合物或其药学上可接受的盐用于治疗过度增殖性疾病例如癌症。
27.在还一方面,本发明涉及如本文提供的式(i)的化合物或其药学上可接受的盐在制备用于治疗过度增殖性疾病例如癌症的药物中的用途。
28.在还一方面,本发明涉及用于治疗过度增殖性疾病例如癌症的试剂盒,该试剂盒包含:如本文提供的式(i)的化合物或其药学上可接受的盐,或包含如本文提供的式(i)的化合物或其药学上可接受的盐和药学上可接受的载体或辅料的药物组合物,容器和任选的指示治疗的包装插页或标签。
具体实施方式
29.现在将详述某些实施方案,其实例在随附的具体实施方式中说明。虽然将描述列举的实施方案,但应理解它们并非旨在将本发明限制于这些实施方案。相反,本发明旨在涵盖所有可选方案、修改和等同方案,它们可以包括在由权利要求限定的本发明的范围内。本领域技术人员将认识到许多与本文所述的方法和材料相似或等同的方法和材料,它们可用于本发明的实践。本发明决不限于所描述的方法和材料。如果一个或更多个并入的文献和
类似材料与本公开不同或相矛盾,包括但不限于定义的术语、术语用法、描述的技术等,以本公开为准。
30.可以理解,本发明的某些特征,为了清楚,在单独实施方案的背景下进行描述,也可以在单个实施方案中组合提供。相反,为了简洁,在单个实施方案的背景下描述的本发明的各种特征也可以单独提供或以任何合适的子组合提供。
31.定义本文使用但未定义的术语具有其通常含义,并且此类术语的含义在其每次出现时均是独立的。然而,除非另有说明,否则以下定义适用于整个说明书和权利要求。
32.如本文所用,术语“包含”和“包括”旨在指定存在所述特征、整体、组件或步骤,但它们不排除存在或添加一个或更多个其他特征、整体、组件、步骤或它们的组。
33.下文更详细地描述了特定官能团和化学术语的定义。出于本发明的目的,化学元素根据元素周期表,cas版,化学和物理手册,第75版,内封面进行鉴定,并且特定官能团通常如其中所述定义。此外,有机化学的一般原理以及特定官能部分和反应性描述于organic chemistry,thomas sorrell,university science books,sausalito,1999;smith和march,march’s advanced organic chemistry,第5版,john wiley & sons,inc.,new york,2001;larock,comprehensive organic transformations,vch publishers,inc.,new york,1989;carruthers,some modem methods of organic synthesis,第3版,cambridge university press,cambridge,1987。
34.除非另有明确说明,否则包含本文引用的所有范围。
35.当列出一系列值时,旨在涵盖该范围内的每个值和子范围。例如,“c
1-6”旨在涵盖c1、c2、c3、c4、c5、c6、c
1-6
、c
1-5
、c
1-4
、c
1-3
、c
1-2
、c
2-6
、c
2-5
、c
2-4
、c
2-3
、c
3-6
、c
3-5
、c
3-4
、c
4-6
、c
4-5
和c
5-6
。
36.当任何变量在任何成分中或在式(i)中或在描绘和描述本发明的化合物的任何其他式中出现多于一次时,它在每次出现时的定义均独立于它在每隔一次出现时的定义。此外,仅当此类组合产生稳定的化合物时,才允许使用取代基和/或变量的组合。
37.如本文所用,术语“烷基”是指直链或支链饱和烃基。术语“c
i-j
烷基”是指具有i至j个碳原子的烷基。除非另有说明,否则烷基可以含有1至10个碳原子。在某些实施方案中,烷基含有1至6个碳原子,例如1至5个碳原子、1至4个碳原子、1至3个碳原子或1至2个碳原子。烷基的非限制性实例包括甲基、乙基、正丙基和异丙基、正丁基、仲丁基、异丁基和叔丁基、新戊基等。
38.如本文所用,术语“单环”是指以单个闭环形式排列的饱和或不饱和的环结构。术语“m元单环”是指具有m个成环原子的单环。除非另有说明,否则单环可以含有3至8个成环原子。在某些实施方案中,单环可以含有3至6个成环原子,例如3个、4个、5个或6个成环原子。在某些实施方案中,作为成环原子,可以是碳原子,也可以是杂原子。在某些实施方案中,单环结构可进一步被取代基团取代。
39.如本文所用,术语“稠环”是包含两个或更多个单环的多环结构,其中,相邻的两个单环共用两个成环原子。包含在稠环中的单环可以是饱和的或不饱和的,可以是芳香性的或非芳香性的。除非另有说明,否则稠环可以含有8至14个成环原子。在某些实施方案中,稠环可以含有8至10个成环原子,例如8个成环原子、9个成环原子或10个成环原子。在某些实施方案中,作为成环原子,可以是碳原子,也可以是杂原子。在某些实施方案中,稠环结构可
进一步被取代基团取代。稠环的非限制性实例包括或。
40.如本文所用,术语“氧代”是指二价氧原子并且氧代的结构可以显示为=o。
41.如本文所用,术语“卤代”或“卤素”是指氟(f)、氯(cl)、溴(br)和碘(i)。在某些实施方案中,卤代的非限制性实例包括氟、氯和溴,更特别地是氟和氯。
42.如本文所用,术语“杂原子”是指氮(n)、氧(o)和硫(s),并且可以包括氮和硫的任何氧化形式,以及碱性氮的任何季铵化形式,除非另有说明。
43.如本文所用,术语“取代的”在指化学基团时是指该化学基团具有一个或更多个氢原子,该氢原子被取代基除去和替换。如本文所用,术语“取代基”具有本领域已知的普通含义并且是指共价附接至母基团或如果合适,稠合至母基团的化学部分。可以理解,给定原子的取代受化合价的限制。应理解取代基可以被进一步取代。
44.当在式(i)或其任何实施方案中指出某部分被“任选地”取代时,这表示式(i)或其实施方案涵盖在该部分上被所指出的取代基取代的化合物和在该部分上不含有所指出的取代基(即,其中该部分未被取代)的化合物。
45.参考通式和具体化合物描述本文提供的化合物。此外,本发明的化合物可以以多种不同的形式或衍生物存在,所有均在本发明的范围内。这些包括例如药学上可接受的盐、互变异构体、立体异构体、外消旋混合物、位置异构体、前药、溶剂化形式、不同晶型或多晶型物以及活性代谢物等。
46.如本文所用,除非另有说明,否则术语“药学上可接受的盐”包括保持特定化合物的游离酸/碱形式的生物学有效性并且在生物学或其他方面不是非期望的盐。药学上可接受的盐可以包括与无机碱或酸和有机碱或酸形成的盐。在本发明的化合物含有一个或更多个酸性或碱性基团的情况下,本发明还包括它们相应的药学上可接受的盐。因此,含有酸性基团(例如羧基基团)的本发明的化合物可以盐形式存在,并且可以根据本发明使用,例如,碱金属盐、碱土金属盐、铝盐或铵盐。此类盐的更多非限制性实例包括锂盐、钠盐、钾盐、钙盐、镁盐、钡盐或与氨或有机胺(例如乙胺、乙醇胺、二乙醇胺、三乙醇胺、哌啶、n-甲基谷氨酰胺或氨基酸)的盐。例如,通过使具有酸性基团的化合物与合适的碱(例如氢氧化锂、氢氧化钠、丙醇钠、氢氧化钾、乙醇钾、氢氧化镁、氢氧化钙或氢氧化钡)反应,这些盐是容易获得的。本发明的化合物的其他碱盐包括但不限于铜(i)、铜(ii)、铁(ii)、铁(iii)、锰(ii)和锌盐。本发明的化合物含有一个或更多个碱性基团,例如可以质子化的基团,可以以盐的形式存在,并且可以根据本发明以它们与无机酸或有机酸的加成盐的形式使用。合适的酸的实例包括氯化氢、溴化氢、碘化氢、磷酸、硫酸、硝酸、甲磺酸、对甲苯磺酸、萘二磺酸、磺基乙酸、三氟乙酸、草酸、乙酸、酒石酸、乳酸、水杨酸、苯甲酸、碳酸、甲酸、丙酸、新戊酸、二乙基乙酸、丙二酸、琥珀酸、庚二酸、富马酸、丙二酸、马来酸、苹果酸、帕莫酸、扁桃酸、氨基磺酸、苯丙酸、葡糖酸、抗坏血酸、异烟酸、柠檬酸、己二酸、牛磺胆酸、戊二酸、硬脂酸、谷氨酸或天冬氨酸,以及本领域技术人员已知的其他酸。所形成的盐尤其是盐酸盐、氯化物、氢溴酸盐、
溴化物、碘化物、硫酸盐、磷酸盐、甲基磺酸盐(甲磺酸盐)、甲苯磺酸盐、碳酸盐、碳酸氢盐、甲酸盐、乙酸盐、磺基乙酸盐、三氟甲磺酸盐、草酸盐、丙二酸盐、马来酸盐、琥珀酸盐、酒石酸盐、苹果酸盐、帕莫酸盐、扁桃酸盐、富马酸盐、乳酸盐、柠檬酸盐、戊二酸盐、硬脂酸盐、天冬氨酸盐或谷氨酸盐。此外,由本发明的化合物形成的盐的化学计量可以是1的整数倍或非整数倍。
47.含有碱性含氮基团的本发明的化合物可以使用试剂例如c
1-4
卤代烷进行季铵化,例如甲基-、乙基-、异丙基-和叔丁基-氯、-溴和-碘;二c
1-4
烷基硫酸盐,例如硫酸二甲酯、二乙酯和二戊酯;c
10-18
烷基卤化物,例如癸基、十二烷基、月桂基、肉豆蔻基和硬脂基的氯化物、溴化物和碘化物;以及芳基c
1-4
烷基卤化物,例如苄基氯和苯乙基溴。
48.如果本发明的化合物在分子中同时含有酸性基团和碱性基团,则除了上述盐形式外,本发明还包括内盐或甜菜碱(两性离子)。相应的盐可以通过本领域技术人员已知的常规方法获得,例如通过将它们与有机或无机酸或碱在溶剂或分散剂中接触,或通过与其他盐的阴离子交换或阳离子交换。本发明还包括本发明的化合物的所有盐,由于低生理相容性,它们不直接适用于药物,但可用于,例如,作为化学反应的中间体或用于制备药学上可接受的盐。有关更合适的盐的综述,参见stahl和wermuth,药用盐手册:特性、选择和使用(wiley-vch,2002)。
49.式(i)的化合物及其药学上可接受的盐可以以非溶剂化和溶剂化形式存在。如本文所用,术语“溶剂化物”是指包含式(i)的化合物或其药学上可接受的盐和一种或更多种药学上可接受的溶剂分子的分子复合物。例如,当溶剂是水时使用术语“水合物”。
50.式(i)的化合物可以具有一个或更多个手性(不对称)中心。本发明涵盖式(i)的化合物的所有立体异构形式。存在于式(i)的化合物中的不对称中心可以彼此独立地具有(r)或(s)构型。当至手性碳的键在本发明的结构式中被描述为直线时,或当化合物名称在没有手性碳的(r)或(s)手性名称的情况下被描述时,应当理解,每种此类手性碳的(r)和(s)构型以及因此每种对映异构体或非对映异构体及其混合物均包含在该式或名称中。特定立体异构体或其混合物的产生可以在获得此类立体异构体或混合物的实例中进行鉴定,但这绝不限制所有立体异构体及其混合物被包括在本发明的范围内。
51.本发明包括所有可能的对映异构体和非对映异构体以及两种或更多种立体异构体的混合物,例如所有比例的对映异构体和/或非对映异构体的混合物。因此,对映异构体是本发明主题的对映异构体纯形式(作为左旋和右旋对映体)、外消旋体形式和两种对映异构体以所有比例的混合物形式。在顺式/反式异构体的情况下,本发明包括顺式形式和反式形式以及这些形式的所有比例的混合物。如有需要,可以通过常规方法(例如通过色谱或结晶、通过使用立体化学均一的合成起始材料或通过立体选择性合成)分离混合物来制备单个立体异构体。任选地,可以在立体异构体分离之前进行衍生化。立体异构体混合物的分离可以在式(i)的化合物合成期间的中间步骤中进行,或者可以在最终外消旋产物上进行。绝对立体化学可以通过结晶产物或结晶中间体的x-射线晶体学来确定,如有需要,用含有已知构型的立体中心的试剂衍生这些结晶产物或结晶中间体。可选地,绝对立体化学可以通过振动圆二色性(vcd)光谱分析来确定。
52.除非另有说明,否则本文描述的结构还意在包括仅在存在一种或更多种同位素富集原子时不同的化合物,换言之,其中一个或更多个原子被具有相同原子序数的原子替换,
但原子质量或质量序数不同于自然界中占优势的原子质量或质量序数的化合物。此类化合物被称为“同位素变体”。本发明旨在包括式(i)的化合物的所有药学上可接受的同位素变体。适合包括在本发明的化合物中的同位素的实例包括但不限于氢的同位素,例如2h(即,d)和3h;碳,例如
11
c、
13
c和
14
c;氯,例如
36
cl;氟,例如
18
f;碘,例如
123
i和
125
i;氮,例如
13
n和
15
n;氧,例如
15
o、
17
o和
18
o;磷,例如
32
p;和硫,例如
35
s。式(i)的化合物的某些同位素变体,例如掺入放射性同位素的那些,可用于药物和/或底物组织分布研究。特别地,具有仅在用较重同位素替换(例如用氘(2h或d)替换氢)中不同的所描绘结构的化合物可以提供某些治疗优势,例如,由于更高的代谢稳定性、增加的体内半衰期或减少的剂量要求,因此可以在一些特定情况下使用。式(i)的化合物的同位素变体通常可以通过本领域技术人员已知的常规技术或通过与所附实施例中描述的那些类似的方法以及使用适当的同位素标记试剂代替先前采用的非标记试剂合成进行制备。
53.根据本发明的药学上可接受的溶剂化物可以包括其中结晶溶剂可以被同位素取代的那些,例如d2o、d
6-丙酮、d
6-dmso。
54.实施本发明的一种方式是以前药的形式施用式(i)的化合物。因此,式(i)的化合物的某些衍生物可能本身具有很少或没有药理活性,当施用于体内或身体上时,例如通过水解切割,特别是由酯酶或肽酶促进的水解切割,其被转化成具有期望活性的式(i)的化合物。此类衍生物被称为“前药”。有关前药使用的更多信息可见于,例如,t. higuchi和w. stella,“pro-drugs as novel delivery systems”,vol. 14,acs symposium series,和e. b. roche(ed.),“bioreversible carriers in drug design”,pergamon press,1987,american pharmaceutical association。还可以参考nature reviews/drug discovery,2008,7,355,和current opinion in drug discovery and development,2007,10,550。
55.根据本发明的前药可以例如通过用本领域技术人员已知的某些部分替换式(i)的化合物中存在的适当官能团来制备,例如,h. bundgaard,“design of prodrugs”,elsevier,1985,以及y. m. choi-sledeski和c. g. wermuth,“designing prodrugs and bioprecursors”,practice of medicinal chemistry,第4版,chapter 28,657-696,elsevier,2015中所述的“前部分”。因此,根据本发明的前药可以包括但不限于(a)式(i)的化合物中羧酸的酯或酰胺衍生物,如果有;(b)式(i)的化合物中氨基的酰胺、亚胺、氨基甲酸酯或胺衍生物;(c)式(i)的化合物中羰基的肟或亚胺衍生物,如果有;或(d)可以在式(i)的化合物中代谢氧化成羧酸的甲基、伯醇或醛基,如果有。
56.提及式(i)的化合物包括化合物本身及其前药。本发明包括此类式(i)的化合物以及此类化合物的药学上可接受的盐和所述化合物和盐的药学上可接受的溶剂化物。
57.使用和施用本发明的化合物
ꢀ‑ꢀ
或其药学上可接受的盐,包括其所有比例的混合物
ꢀ‑ꢀ
可用作药物。该系列的化合物作为细胞周期蛋白依赖性激酶的atp竞争性抑制剂,对cdk2/4的酶水平抑制活性优异,相对于同源性较高的cdk1/9表现出了较好的选择性,脱靶引起的安全性风险小。
58.作为cdk选择性抑制剂的本发明的化合物特别适用于治疗过度增殖性疾病例如癌症,包括但不限于乳腺癌、脑癌、结直肠癌、肺癌、胃癌、肝癌、淋巴瘤黑色素瘤、卵巢癌、胰腺癌和前列腺癌等,特别是结直肠癌、卵巢癌和乳腺癌。
59.如本文所用,术语“过度增殖性疾病”是指受试者体内出现不期望或不受控制的细胞增殖的疾病。在某些实施方案中,过度增殖性疾病是癌症。
60.本发明的化合物可以以有效治疗本文所述的疾病或病状的量施用。本发明的化合物可以作为化合物本身施用,或可选地,作为药学上可接受的盐施用。为了施用和给药目的,本发明的化合物本身或其药学上可接受的盐将被简称为本发明的化合物。
61.本发明的化合物通过任何合适的途径以适合这种途径的药物组合物的形式施用,并且以对预期治疗有效的剂量施用。本发明的化合物可以口服、直肠、阴道、肠胃外或外用施用。
62.如本文所用,术语“施用”是指吸收、摄取、注射、吸入、植入或以其他方式引入本发明的化合物或其药物组合物。术语“治疗”是指逆转、减轻、延迟本文所述的“病理状况”(例如,疾病、病症或病状,或其一种或更多种体征或症状)的发作或抑制其进展。在某些实施方案中,可以在疾病或病状的一种或更多种体征或症状已经发展或已经观察到之后施用治疗。在其他实施方案中,可以在没有疾病或病状的体征或症状的情况下进行治疗。例如,可以在症状发作之前对易感个体进行治疗(例如,根据症状史和/或根据遗传或其他易感性因素)。在症状消退后也可以继续治疗,例如延迟或预防复发。如本文所用,术语“疾病”、“病症”、“病状”和“病理状况”可互换使用。
63.本领域技术人员可以通过常规实验确定施用的剂量水平。本发明的化合物和/或包含所述化合物的组合物的剂量方案基于多种因素,包括患者的类型、年龄、体重、性别和医疗状况;病状的严重程度;施用途径;和所用特定化合物的活性。因此,剂量方案可以有很大的不同。例如,本发明的化合物的剂量水平可以是每天约0.001至约100 mg/kg(即mg/kg体重)。在某些实施方案中,以单次或分次施用的本发明的化合物的总日剂量可以为约0.001至约10 mg/kg。本发明的化合物的施用可以在一天内重复多次的情况并不少见。
64.药物组合物在一些方面,本发明涉及药物组合物,其包含如本文提供的式(i)的化合物或其药学上可接受的盐,以及至少一种药学上可接受的载体或辅料。
65.如本文所用,术语“药学上可接受的载体或辅料”是指可用于制备药物组合物的载体或辅料,其通常是安全的、无毒的并且在生物学上或其他方面均不是有不利影响的,并且包括可接受用于兽医用途以及人药物用途的载体或辅料。如本文所用的药学上可接受的载体或辅料包括一种和多于一种此类载体或辅料。使用的具体载体或辅料将取决于应用本发明的化合物的方式和目的。合适的载体和辅料是本领域技术人员所熟知,并详述于,例如,ansel,howard c等人,ansel’s pharmaceutical dosage forms and drug delivery systems. philadelphia:lippincott,williams & wilkins,2004;gennaro,alfonso r.等人,remington:the science and practice of pharmacy. philadelphia:lippincott,williams & wilkins,2000;和rowe,raymond c. handbook of pharmaceutical excipients. chicago,pharmaceutical press,2005。还可以包括缓冲剂、稳定剂、表面活性剂、润湿剂、润滑剂、乳化剂、混悬剂、防腐剂、抗氧化剂、遮光剂、助流剂、加工助剂、着色剂、甜味剂、加香剂、调味剂、稀释剂和其他已知的添加剂中的一种或更多种,以提供药物(即本文提供的化合物或药物组合物)的精致外观表现或有助于生产药物产品(即药物)。
66.本发明的组合物可以制剂成多种形式。这些包括,例如,液体、半固体和固体剂型,
例如液体溶液(例如,可注射和可输注的溶液)、分散体或混悬剂、片剂、丸剂、粉剂、脂质体、栓剂等。形式取决于预期施用的方式和治疗应用。
67.本发明的药物组合物可以通过任何熟知的药学技术(例如有效制剂和施用程序)制备。上述关于有效制剂和施用程序的考虑是本领域熟知的,并且在标准教科书中有所描述。例如,在hoover,john e.,remington’s pharmaceutical sciences,mack publishing co.,easton,pennsylvania,1975;liberman等人,编辑,pharmaceutical dosage forms,marcel decker,new york,n.y.,1980;和kibbe等人,编辑,handbook of pharmaceutical excipients,第3版,american pharmaceutical association,washington,1999中讨论了药物产品的制剂。
68.在还一方面,本发明涉及用于治疗过度增殖性疾病例如癌症的试剂盒,其包含如本文提供的式(i)的化合物或其药学上可接受的盐,或如本文提供的包含式(i)的化合物或其药学上可接受的盐的药物组合物,容器和任选的指示治疗的包装插页或标签。
69.治疗方法在还一方面,本发明涉及一种治疗有需要的受试者的过度增殖性疾病例如癌症的方法,由于本发明的化合物的cdk抑制活性,该方法包括向受试者施用治疗有效量的如本文提供的式(i)的化合物或其药学上可接受的盐。
70.如本文所用,术语“有需要的受试者”是患有过度增殖性疾病例如癌症的受试者,或相对于整个人群发展过度增殖性疾病例如癌症的风险增大的受试者。在某些实施方案中,受试者是温血动物。在某些实施方案中,温血动物是哺乳动物。在某些实施方案中,温血动物是人。
71.在某些实施方案中,癌症包括选自以下的任一种:乳腺癌、脑癌、结直肠癌、肺癌、胃癌、肝癌、淋巴瘤黑色素瘤、卵巢癌、胰腺癌和前列腺癌等,特别是结直肠癌、卵巢癌和乳腺癌。
72.如本文所述的治疗过度增殖性疾病例如癌症的方法可以用作单一疗法。如本文所用,术语“单一疗法”是指向有需要的受试者施用单一活性或治疗性化合物。在某些实施方案中,单一疗法将涉及向需要这种治疗的受试者施用治疗有效量的一种本发明的化合物或其药学上可接受的盐。
73.在还一方面,本发明涉及如本文提供的式(i)的化合物或其药学上可接受的盐用于治疗过度增殖性疾病例如癌症。
74.在还一方面,本发明涉及如本文提供的式(i)的化合物或其药学上可接受的盐在制备用于治疗过度增殖性疾病例如癌症的药物中的用途。
75.合成本发明的化合物可以使用合成有机化学领域技术人员的公知常识,通过下文描述的一般和特定方法制备。这种公知常识可见于标准参考书,例如,barton和ollis(编辑),《综合有机化学》,elsevier;richard larock,《综合有机转化:官能团制备指南》,john wiley and sons;和《有机合成方法纲要》,i-xii卷,wiley-interscience。
76.下文描述的方案旨在提供用于制备本发明的化合物的方法的一般描述。本发明的一些化合物可以含有单个或多个具有立体化学名称(r)或(s)的手性中心。对本领域技术人员显而易见的是,无论材料是对映体富集的还是外消旋的,所有合成转化均可以以类似的
方式进行。此外,期望的光学活性材料的解析可以使用公知的方法,例如本文和化学文献中描述的那些方法,在程序中的任何期望点进行。
77.实施例为了更详细地描述本发明,提出以下实施例。本文描述的实施例用于说明本文提供的化合物、方法和组合物,并且不应以任何方式解释为限制它们的范围。
78.在合成过程中,可能需要和/或期望保护任何相关分子上的敏感或反应性基团。这可以通过常规保护基团来实现,例如在t.w. greene和p.g.m. wutts,《有机合成中的保护基团》,第4版,john wiley and sons中所述的那些保护基团。使用本领域熟知的方法在方便的后续阶段任选地去除保护基团。
79.本发明的化合物可以根据以下反应方案和实施例或其修改,使用容易获得的起始材料、试剂和常规合成程序容易地制备。在这些反应中,也可以使用本领域技术人员已知但未更详细提及的变体。此外,根据本文所述的反应方案和实施例,制备本发明的化合物的其他方法对于本领域技术人员将是显而易见的。除非另有说明,否则所有变量均如上定义。一般而言,化学程序中,所有试剂和起始材料均可购自商业供应商或可由本领域技术人员容易地制备。
80.本发明所用的缩写定义如下:pd(dppf)cl2代表1,1'-二(二苯膦基)二茂铁二氯化钯(ii);ruphos代表2-二环己基磷-2',6'-二异丙氧基-1,1'-联苯;pd2(dba)3代表三(二亚苄基丙酮)二钯;dmso代表二甲基亚砜;hepes代表4-(2-羟乙基)-1-哌嗪乙磺酸;dtt代表二硫苏糖醇;atp代表腺嘌呤核苷三磷酸;dyrk代表双特异性酪氨酸磷酸化调节激酶。
81.实施例1:步骤1:式(1-2)所示化合物的合成将式(1-1)所示的化合物溶于10毫升四氢呋喃中。随后,在室温下向其中加入3,4-二氢-2h-吡喃(0.80 g,9.5 mmol)和对甲苯磺酸(0.08 g,0.48 mmol)。60℃下反应2小时,反应结束后,减压蒸除溶剂,残余物通过柱层析分离后得到式(1-2)所示化合物(700 mg,2.4 mmol)。1h nmr(400 mhz,cdcl3)δ 7.60(s,1h),5.99(dd,j = 10.6,1.9 hz,1h),4.20
ꢀ‑ꢀ
4.05(m,1h),3.73(td,j = 11.4,2.4 hz,1h),2.22
ꢀ‑ꢀ
2.10(m,1h),2.05(s,1h),1.78
ꢀ‑ꢀ
1.60(m,4h)。
82.步骤2:式(1-4)所示化合物的的合成将式(1-3)所示的化合物(800 mg,2.5 mmol)溶于12毫升四氢呋喃和2.5毫升水的混合溶液中。随后在室温和氮气氛围下,向其中加入式(1-2)所示化合物(731.1 mg,2.5 mmol),碳酸钾(688.4 mg,5.0 mmol)和pd(dppf)cl2(182.2 mg,0.25 mmol)。90℃下反应3小时。反应结束后,减压蒸除溶剂,残余物通过柱层析分离后得到式(1-4)所示化合物。lcms:408.1 [m+h]
+
。
[0083]
步骤3:式(1-6)所示化合物的合成将式(1-4)所示化合物(400 mg,0.98 mmol)和式(1-5)所示化合物(186 mg,0.98 mmol)溶于10毫升二氧六环中。随后在室温和氮气氛围下,向体系中加入碳酸铯(639.1 mg,1.96 mmol),ruphos(137.3 mg,0.29 mmol)和pd2(dba)3(89.81 mg,0.098 mmol)。90℃下反应6小时。反应结束后,减压蒸除溶剂,残余物通过柱层析分离后得到式(1-6)所示化合物。lcms:562.5 [m+h]
+
。
[0084]
步骤4:式(1)所示化合物的合成将式(1-6)所示化合物(330 mg,0.59 mmol)溶于10毫升二氧六环和5毫升氯化氢甲醇溶液中。室温下反应1小时。反应结束后,减压蒸除溶剂,残余物通过制备高效液相色谱分离后得到式(1)所示化合物。lcms:478.0 [m+h]
+
;1h nmr(400 mhz,dmso-d6)δ 12.12(s,1h),8.87(s,1h),7.49(d,j = 8.6 hz,2h),7.35(s,1h),7.23
ꢀ‑ꢀ
6.93(m,4h),4.33
ꢀ‑ꢀ
4.21(m,2h),4.15(p,j = 6.6 hz,1h),3.35
ꢀ‑ꢀ
3.25(m,2h),3.24
ꢀ‑ꢀ
3.06(m,2h),2.61
ꢀ‑ꢀ
2.51(m,3h),2.50(s,3h),1.95
ꢀ‑ꢀ
1.60(m,4h),1.17(d,j = 6.5 hz,6h)。
[0085]
实施例2:参考实施例1中的步骤3和步骤4,以式(2-1)所示化合物与式(1-4)所示化合物为原料,同法制得式(2)所示化合物。lcms:479.3 [m+h]
+
;1h nmr(400 mhz,dmso-d6)δ 12.31(s,1h),9.36(s,1h),8.31(s,1h),8.09(s,1h),7.75(s,1h),7.61(s,2h),7.18
ꢀ‑ꢀ
7.07(m,1h),4.29
ꢀ‑ꢀ
4.21(m,2h),4.15
ꢀ‑ꢀ
4.06(m,1h),3.29
ꢀ‑ꢀ
3.25(m,2h),2.92
ꢀ‑ꢀ
2.84(m,2h),2.45
ꢀ‑ꢀ
2.39(m,1h),2.21(s,3h),1.99(t,j = 10.4 hz,2h),1.77
ꢀ‑ꢀ
1.59(m,4h),1.16(d,j = 6.5 hz,6h)。
[0086]
实施例3:
步骤1:式(3-3)所示化合物的合成将式(3-2)所示化合物(315.81 mg,2.46 mmol)与碳酸钾(1021.18 mg,7.39 mmol)溶于5毫升n,n-二甲基甲酰胺。随后在室温氮气氛围下,向其中加入式(3-1)所示化合物(500 mg,2.46 mmol)。80℃下反应12小时。反应结束后,向反应体系中加入20毫升水进行稀释。使用30毫升乙酸乙酯对所得混合物进行萃取,重复三次。将合并后的有机相溶液依次使用饱和食盐水洗涤,无水硫酸钠进行干燥,过滤除去硫酸钠,减压蒸除溶剂,残余物通过制备柱层析分离后得到式(3-3)所示化合物。lcms:251.0 [m+h]
+
。
[0087]
步骤2:式(3-4)所示化合物的合成将式(3-3)所示化合物(200 mg,0.8 mmol)溶于10毫升甲醇中。随后在室温和氮气氛围下,向其中加入pd/c(10%,170.1 mg,0.16 mmol)。使用氢气对反应体系进行抽换气操作三次。随后在15 psi压力的氢气氛围和室温下反应4小时。反应结束后,通过硅藻土过滤除去体系中的pd/c,减压蒸除溶剂,不必经过进一步纯化,即可获得式(3-4)所示化合物。lcms:221.2 [m+h]
+
。
[0088]
步骤3:式(3)所示化合物的合成参考实施例1中的步骤3和步骤4,以式(3-4)所示化合物与式(1-4)所示化合物为原料,同法制得式(3)所示化合物。lcms:508.3 [m+h]
+
;1h nmr(400 mhz,dmso-d6)δ 12.20(s,1h),9.13(s,1h),8.30(s,1h),7.93(d,j = 2.8 hz,1h),7.66(s,1h),7.65
ꢀ‑ꢀ
7.57(m,1h),7.39(dd,j = 9.2,3.0 hz,1h),7.14
ꢀ‑ꢀ
7.03(m,1h),4.33
ꢀ‑ꢀ
4.19(m,2h),4.18
ꢀ‑ꢀ
4.04(m,1h),3.62
ꢀ‑ꢀ
3.54(m,2h),2.68
ꢀ‑ꢀ
2.58(m,2h),2.36
ꢀ‑ꢀ
2.29(m,1h),2.20(s,6h),2.19
ꢀ‑ꢀ
2.10(m,2h),1.90
ꢀ‑ꢀ
1.76(m,2h),1.55
ꢀ‑ꢀ
1.41(m,2h),1.16(d,j = 6.6 hz,6h)。
[0089]
实施例4:
步骤1:式(4-3)所示化合物的合成将式(4-1)所示化合物(3 g,26.5 mmol)溶于50毫升的二氯甲烷和20毫升四氢呋喃的混合溶液中。随后,向溶液中依次加入式(4-2)所示化合物(6.0 g,106.1 mmol),醋酸(2.3 ml,39.8 mmol)和三乙酰氧基硼氢化钠(11.24 g,53.041 mmol)。40℃下反应过夜。反应结束后,减压蒸除溶剂,残余物通过柱层析分离后得到式(4-3)所示化合物。lcms:141.2 [m+h]
+
。
[0090]
步骤2:式(4-4)所示化合物的合成将式(4-3)所示化合物(0.4 g,2.85 mmol)和式(3-1)所示化合物(0.87 g,4.28 mmol)溶于10毫升二氧六环中。随后,向其中加入碳酸铯(1.86 g,5.71 mmol),ruphos(0.27 g,0.57 mmol)和pd2(dba)3(0.26 g,0.285 mmol)。在氮气氛围和100℃下反应3小时。反应结束后,减压蒸除溶剂,残余物通过柱层析分离后得到式(4-4)所示化合物。lcms:263.0 [m+h]
+
。
[0091]
步骤3:式(4-5)所示化合物的合成参考实施例3中的步骤2,以式(4-4)所示化合物为原料,同法制得式(4-5)所示化合物。lcms:235.2 [m+h]
+
。
[0092]
步骤4:式(4)所示化合物的合成参考实施例1中的步骤3和步骤4,以式(4-5)所示化合物与式(1-4)所示化合物为原料,同法制得式(4)所示化合物。1h nmr(400 mhz,dmso-d6)δ 12.36(s,1h),9.55(s,1h),8.15
ꢀ‑ꢀ
8.13(m,1h),7.74(s,1h),7.71
ꢀ‑ꢀ
7.67(m,1h),7.63(dd,j = 8.9,2.6 hz,1h),7.14(s,1h),7.11(dd,j = 11.9,1.8 hz,1h),4.30
ꢀ‑ꢀ
4.22(m,2h),4.12(p,j = 6.7 hz,1h),3.68
ꢀ‑ꢀ
3.57(m,2h),3.29
ꢀ‑ꢀ
3.26(m,2h),2.70
ꢀ‑ꢀ
2.59(m,2h),2.40(s,6h),2.34
ꢀ‑ꢀ
2.32(m,1h),2.21
ꢀ‑ꢀ
2.10(m,1h),1.93
ꢀ‑ꢀ
1.77(m,1h),1.17(d,j = 6.6 hz,6h)。
[0093]
实施例5:
步骤1:式(5-2)的合成将无水三氯化铝(3.0 g,22.8 mmol)均匀悬浮于25毫升二氯甲烷后,在0℃和氮气氛围下向该混合物中加入2-溴-2-甲基丙烷(1.25 g,9.1 mmol)。继续在0℃下搅拌10分钟后,向体系中加入式(5-1)所示化合物(1.5 g,7.6 mmol)。0℃下反应3小时后,向体系中加入30毫升冰水进行淬灭。使用50毫升二氯甲烷对该体系进行萃取,重复三次。合并后的有机相经50毫升饱和食盐水洗涤和无水硫酸钠干燥后,过滤,减压蒸除溶剂,残余物通过柱层析分离后得到式(5-2)所示化合物。lcms:253.1[m+h]
+
。
[0094]
步骤2:式(5-3)所示化合物的合成将式(5-2)所示化合物(260 mg,1.03 mmol),4,4,5,5-四甲基-2-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊-2-基)-1,3,2-二氧杂硼杂环戊烷(782.47 mg,3.08 mmol)和醋酸钾(302.4 mg,3.08 mmol)溶于10毫升二氧六环。随后,向该体系中加入pd(dppf)cl2(150.3 mg,0.205 mmol)。90℃和氮气氛围下反应12小时后,经过硅藻土过滤,减压蒸除溶剂,残余物通过柱层析分离后得到式(5-3)所示化合物。lcms:301.1 [m+h]
+
。
[0095]
步骤3:式(5-4)所示化合物的合成将式(5-3)所示化合物(196.4 mg,0.65 mmol),式(1-2)所示化合物(160 mg,0.55 mmol),碳酸钾(150.6 mg,1.09 mmol)和pd(dppf)cl2(39.88 mg,0.055 mmol)溶于3毫升二氧六环和1.5毫升水的混合溶剂中。在90℃和氮气氛围下反应12小时后,向体系中加入20毫升水和40毫升乙酸乙酯,然后进行分液,有机相经20毫升饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压蒸除溶剂,残余物通过柱层析分离后得到式(5-4)所示化合物。lcms:387.1 [m+h]
+
。
[0096]
步骤4:式(5)所示化合物的合成参考实施例1中的步骤3和步骤4,以式(5-4)所示化合物和式(2-1)所示化合物为原料,同法制得式(5)所示化合物。lcms:458.2 [m+h]
+
;1h nmr(400 mhz,dmso-d6)δ 12.48(s,1h),9.45(s,1h),8.69(d,j = 7.4 hz,2h),8.11(s,1h),7.99(s,1h),7.89(s,1h),7.63(s,2h),7.20(dd,j = 7.3,1.9 hz,1h),2.94(d,j = 11.2 hz,2h),2.48
ꢀ‑ꢀ
2.40(m,1h),2.27(s,3h),2.16
ꢀ‑ꢀ
2.00(m,2h),1.83
ꢀ‑ꢀ
1.59(m,4h),1.44(s,9h)。
[0097]
生物活性评价以下实施例证明了本发明的化合物的生物活性。
[0098]
cdk1将上述实施例制备的化合物1-5使用dmso进行稀释,稀释至检测浓度的200
×
。用echo 665转移20 nl的化合物至384反应板中(784075,greiner)。用1
×
的激酶反应缓冲液(50 mm hepes,10 mm mgcl2,0.01% brij,2 mm dtt),准备2
×
的激酶溶液,转移2 μl的激酶溶液到384反应板中。使用离心机在1000 rpm离心1分钟,25℃孵育10分钟。用激酶反应缓冲液准备2
×
的底物(h1:0.1 mg/ml)和atp(50 μm)的混合液,向反应板中加入2 μl的底物和atp的混合液用来开始反应,使用离心机在1000 rpm离心1分钟。使用封板膜封住反应板,25℃孵育60分钟。向反应板的每个孔中加入4 μl室温孵育的adp-glo试剂(promega,cat:v9103),1000 rpm离心1分钟,25℃孵育40分钟。向反应板的每个孔中加入8 μl室温孵育的检测试剂(promega,cat:v9103),1000 rpm离心1分钟,25℃孵育40分钟。使用bmg读取化学发光(luminescence)信号。
[0099]
抑制率计算如下:化合物抑制率(% inh)= 100%
ꢀ‑
(化合物-阳性对照)/(阴性对照-阳性对照)
×
100%。
[0100]
ic
50
通过抑制率由prism graphpad7.0计算。
[0101]
cdk2将上述实施例制备的化合物1-5使用dmso进行稀释,稀释至检测浓度的200
×
。用echo 665转移20 nl的化合物至384反应板中(784075,greiner)。用1
×
的激酶反应缓冲液(50 mm hepes,10 mm mgcl2,0.01% brij,2 mm dtt),准备2
×
的激酶溶液,转移2 μl的激酶溶液到384反应板中。使用离心机在1000 rpm离心1分钟,25℃孵育10分钟。用激酶反应缓冲液准备2
×
的底物(h1:0.1 mg/ml)和atp(20 μm)的混合液,向反应板中加入2 μl的底物和atp的混合液用来开始反应,使用离心机在1000 rpm离心1分钟。使用封板膜封住反应板,25℃孵育60分钟。向反应板的每个孔中加入4 μl室温孵育的adp-glo试剂(promega,cat:v9103),1000 rpm离心1分钟,25℃孵育40分钟。向反应板的每个孔中加入8 μl室温孵育的检测试剂(promega,cat:v9103),1000 rpm离心1分钟,25℃孵育40分钟。使用bmg读取化学发光(luminescence)信号。
[0102]
抑制率计算如下:化合物抑制率(% inh)= 100%
ꢀ‑
(化合物-阳性对照)/(阴性对照-阳性对照)
×
100%。
[0103]
ic
50
通过抑制率由prism graphpad7.0计算。
[0104]
cdk4将上述实施例制备的化合物1-5使用dmso进行稀释,稀释至检测浓度的200
×
。用echo 665转移20nl的化合物至384反应板中(784075,greiner)。用1
×
的激酶反应缓冲液(50 mm hepes,10 mm mgcl2,0.01% brij,2 mm dtt),准备2
×
的激酶溶液,转移2 μl的激酶溶液到384反应板中。使用离心机在1000 rpm离心1分钟,25℃孵育10分钟。用激酶反应缓冲液准备2
×
的底物(dyrk:0.1 mg/ml)和atp(100 μm)的混合液,向反应板中加入2 μl的底物和atp的混合液用来开始反应,使用离心机在1000 rpm离心1分钟。使用封板膜封住反应板,25℃孵育60分钟。向反应板的每个孔中加入4 μl室温孵育的adp-glo试剂(promega,cat:v9103),1000 rpm离心1分钟,25℃孵育40分钟。向反应板的每个孔中加入8 μl室温孵
育的检测试剂(promega,cat:v9103),1000 rpm离心1分钟,25℃孵育40分钟。使用bmg读取化学发光(luminescence)信号。
[0105]
抑制率计算如下:化合物抑制率(% inh)= 100%
ꢀ‑
(化合物-阳性对照)/(阴性对照-阳性对照)
×
100%。
[0106]
ic
50
通过抑制率由prism graphpad7.0计算。
[0107]
cdk9将上述实施例制备的化合物1-5使用dmso进行稀释,稀释至检测浓度的200
×
。用echo 665转移20 nl的化合物至384反应板中(784075,greiner)。用1
×
的激酶反应缓冲液(50mm hepes,10mm mgcl2,0.01% brij,2mm dtt),准备2
×
的激酶溶液,转移2 μl的激酶溶液到384反应板中。使用离心机在1000 rpm离心1分钟,25℃孵育10分钟。用激酶反应缓冲液准备2
×
的底物(cdk:0.1 mg/ml)和atp(20 μm)的混合液,向反应板中加入2 μl的底物和atp的混合液用来开始反应,使用离心机在1000 rpm离心1分钟。使用封板膜封住反应板,25℃孵育60分钟。向反应板的每个孔中加入4 μl室温孵育的adp-glo试剂(promega,cat:v9103),1000 rpm离心1分钟,25℃孵育40分钟。向反应板的每个孔中加入8 μl室温孵育的检测试剂(promega,cat:v9103),1000 rpm离心1分钟,25℃孵育40分钟。使用bmg读取化学发光(luminescence)信号。
[0108]
抑制率计算如下:化合物抑制率(% inh)= 100%
ꢀ‑
(化合物-阳性对照)/(阴性对照-阳性对照)
×
100%。
[0109]
ic
50
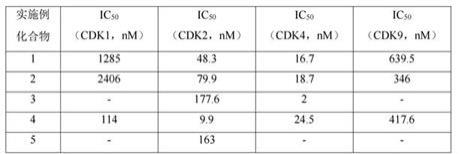
通过抑制率由prism graphpad7.0计算。
[0110]
上述实验得到的本发明的化合物1-5针对cdk的ic
50
总结在以下表1中:表1注:
“‑”
表示未检测由上表可知,本发明的化合物对cdk2/4具有较高的选择性,能够有效地抑制cdk2/4的活性。
[0111]
前面的描述被认为仅是对本发明原理的说明。此外,由于对本领域技术人员而言许多修改和变化将是显而易见的,因此不希望将本发明限制于如上所述的确切构造和过程。因此,所有合适的修改和等同物均可以被认为落入由所附权利要求限定的本发明的范围内。
[0112]
本文引用的所有出版物、专利和专利申请均通过引用以其全文并入本公开。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1