一种治疗成人T细胞白血病淋巴瘤药物伐美妥司他的制备方法与流程
发明领域本发明属于化学药物领域,具体涉及一种复发或难治性成人t细胞白血病-淋巴瘤治疗药物伐美妥司他(valemetostat)新的制备方法。
背景技术:
1、成人t细胞白血病-淋巴瘤(atl)是一种外周t细胞肿瘤,分为急性型、淋巴瘤型和具有不良预后因素的慢性型。复发或难治性atl仍是一种预后较差的疾病。在atl细胞中,ezh2表达升高,甲基化组蛋白在大约一半的基因位点异常积累。
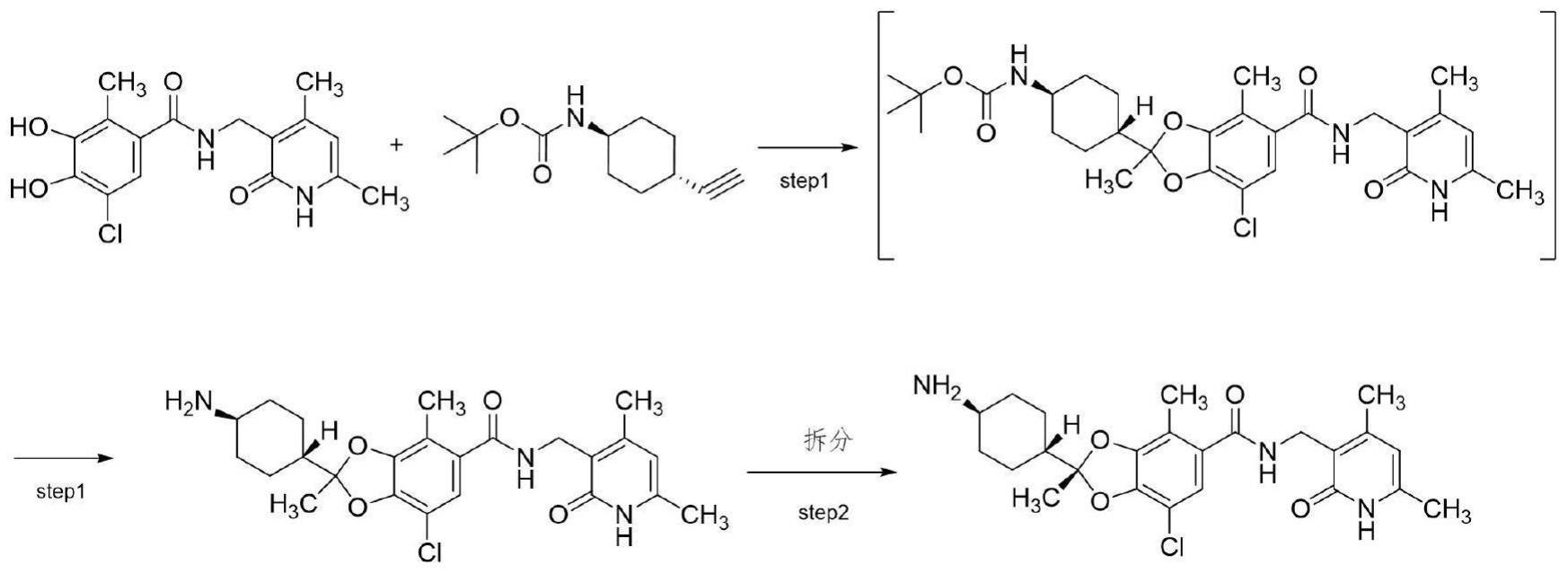
2、ezh1和ezh2是甲基转移酶,可特异性甲基化组蛋白h3的第27个赖氨酸残基(histone h3 lysine 27:h3k27),通过形成prc2(polycomb repressive complex 2)的蛋白质复合物来表现出甲基转移酶活性。prc2复合物有两种类型,prc2-ezh1和prc2-ezh2,其催化亚基分别为ezh1和ezh2。prc2-ezh1和prc2-ezh2均可对h3k27进行单甲基化、二甲基化和三甲基化。
3、单独抑制ezh2可使甲基化组蛋白保持完整,同时抑制ezh1和ezh2可去除异常积累的甲基化组蛋白,显着降低atl细胞系和atl患者的atl细胞的活力。尽管作用机制不详细,但是这种抑制ezh1和ezh2的药物在体外测试系统中显示出比ezh2抑制剂更强的抗atl细胞系增殖活性。h3k27的三甲基化抑制肿瘤抑制基因和分化相关基因的表达,prc2复合体被认为在癌症的发生和发展中起重要作用。
4、伐美妥司他甲苯磺酸盐(valemetostat tosilate)为第一三共株式会社开发的口服抗癌药,伐美妥司他甲苯磺酸盐valemetostat tosilate的中文化学名称:(2r)-7-氯-2-[反式-4-(二甲基氨基)环己基]-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧基-5-甲酰胺单(4-甲基苯磺酸盐),分子式:c26h34cln3o4·c7h8o3s,分子量:660.22,cas登记号:1809336-93-3,其化学结构式如下:
5、
6、伐美妥司他甲苯磺酸盐是enhancer of zeste homolog(ezh1和ezh2)的选择性抑制剂,通过抑制ezh1/2的甲基化活性,抑制h3k27等的甲基化,推测具有抑制肿瘤生长的作用。
7、在伐美妥司他甲苯磺酸盐的一项日本针对复发或难治性atl患者的ii期临床研究中:主要终点反应率(95%ci)为48.0%(27.8-68.7%)。入组患者分病种反应率:急性型62.5%(10/16例),淋巴瘤型16.7%(1/6例),具有不良预后因素的慢性型33.3%(1/3例);次要终点中位无进展生存期(95%ci)、32.14周(13.14周至不可估计)、疾病控制率(95%ci)为88.0%(68.8-97.5%)。
8、现有技术文件专利cn201680044712和cn201580014878报道了伐美妥司他的合成路线1如下:
9、
10、步骤1:起始原料5-氯-3,4-二羟基-2-甲基苯甲酸甲酯和n-(反式-4-乙炔基环己基)氨基甲酸叔丁酯反应得到中间体(2rs)-2-[反式-4-(叔丁氧基羰基氨基)环己基]-7-氯-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酸甲酯,通过硅胶柱色谱法纯化。
11、步骤2:上步中间体(2rs)-2-[反式-4-(叔丁氧基羰基氨基)环己基]-7-氯-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酸甲酯通过手性柱色谱拆分得到中间体(2r)-2-[反式-4-(叔丁氧基羰基氨基)环己基]-7-氯-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酸甲酯,收率23%。
12、步骤3:上步中间体(2r)-2-[反式-4-(叔丁氧基羰基氨基)环己基]-7-氯-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酸甲酯通过氢氧化钠水解得到中间体(2r)-2-[反式-4-(叔丁氧基羰基氨基)环己基]-7-氯-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酸。
13、步骤4:上步中间体(2r)-2-[反式-4-(叔丁氧基羰基氨基)环己基]-7-氯-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酸与起始物料3-(氨基甲基)-4,6-二甲基-1,2-二氢吡啶-2-酮盐酸盐发生酰胺化反应得到中间体n-[反式-4-[(2r)-7-氯-5-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基氨基甲酰基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-2-基]环己基]氨基甲酸叔丁酯,通过硅胶柱色谱法纯化。
14、步骤5:上步中间体n-[反式-4-[(2r)-7-氯-5-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基氨基甲酰基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-2-基]环己基]氨基甲酸叔丁酯在盐酸中脱保护基得到中间体(2r)-2-(反式-4-氨基环己基)-7-氯-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酰胺。
15、步骤6:上步中间体(2r)-2-(反式-4-氨基环己基)-7-氯-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酰胺与甲醛发生氮甲基化反应得到(2r)-7-氯-2-[反式-4-(二甲基氨基)环己基]-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酰胺(伐美妥司他),通过硅胶柱色谱法纯化。
16、该路线步骤较长,步骤2采用手性柱拆分收率仅23%,且多步采用柱层析纯化,较难产业化。
17、现有技术文件专利tw202216654a报道了新的伐美妥司他合成路线2,具体如下:
18、
19、步骤1:起始原料3,4-2-羟基-2-甲基苄酸甲酯与硫酰氯反应得到中间体5-氯-3,4-二羟基-2-甲基苯甲酸甲酯;
20、步骤2:上步中间体5-氯-3,4-二羟基-2-甲基苯甲酸甲酯和n-(反式-4-乙炔基环己基)氨基甲酸叔丁酯反应得到中间体(2rs)-2-[反式-4-(叔丁氧基羰基氨基)环己基]-7-氯-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酸甲酯。
21、步骤3:上步中间体(2rs)-2-[反式-4-(叔丁氧基羰基氨基)环己基]-7-氯-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酸甲酯用氢氧化钠水解得到中间体(2rs)-2-[反式-4-(叔丁氧基羰基氨基)环己基]-7-氯-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酸。
22、步骤4:上步中间体(2rs)-2-[反式-4-(叔丁氧基羰基氨基)环己基]-7-氯-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酸通过拆分剂s-1-苯乙胺得到中间体(2r)-2-[反式-4-(叔丁氧基羰基氨基)环己基]-7-氯-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酸的s-1-苯乙胺盐。
23、步骤5:上步中间体(2r)-2-[反式-4-(叔丁氧基羰基氨基)环己基]-7-氯-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酸的s-1-苯乙胺盐经解盐、脱boc保护基及氮甲基化得到中间体(2r)-2-[反式-4-(二甲基氨基)环己基]-7-氯-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酸盐酸盐。
24、步骤6:上步中间体(2r)-2-[反式-4-(二甲基氨基)环己基]-7-氯-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酸盐酸盐与起始物料3-(氨基甲基)-4,6-二甲基-1,2-二氢吡啶-2-酮盐酸盐发生酰胺化反应得到伐美妥司他。
25、上述路线2以3,4-2-羟基-2-甲基苄酸甲酯为起始原料,经6步合成伐美妥司他,其中步骤4采用拆分剂进行手性拆分;拆分后中间体先进行氮甲基化反应,再进行3-(氨基甲基)-4,6-二甲基-1,2-二氢吡啶-2-酮盐酸盐的酰胺对接,虽然整个工艺未使用柱色谱纯化,但仍存在路线较长,收率低的问题。
26、综上,现有技术文件报道的伐美妥司他的合成路线存在路线长,收率低,导致原料药存在可及性差的问题,因此,有必要进行合成工艺的技术创新,来提高原料药的可及性。
技术实现思路
1、本发明的目的在于克服现有技术的不足,提供一种伐美妥司他的制备方法。
2、本发明仅需2步反应得到伐美妥司他中间体(2r)-2-(反式-4-氨基环己基)-7-氯-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酰胺新的制备方法,该中间体再与甲醛发生氮甲基化反应可得到伐美妥司他。
3、本发明所述方法为:
4、
5、步骤1、5-氯-n-((4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基)-3,4-二羟基-2-甲基苯甲酰胺和n-(反式-4-乙炔基环己基)氨基甲酸叔丁酯在非质子溶剂中以钌金属化合物和膦配体化合物催化剂,经c-o交叉偶联反应得到中间态(2rs)-2-[反式-4-(叔丁氧基羰基氨基)环己基]-7-氯-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酰胺,再脱保护基得到(2rs)-2-(反式-4-氨基环己基)-7-氯-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酰胺;
6、步骤2、(2rs)-2-(反式-4-氨基环己基)-7-氯-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酰胺通过拆分得到单一构型(2r)-2-(反式-4-氨基环己基)-7-氯-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酰胺。
7、本发明路线不同于现有技术文件专利cn201680044712和cn201580014878报道的伐美妥司他的合成路线1,也不同于现有技术文件专利tw202216654a报道的伐美妥司他合成路线2。
8、本发明通过2步操作制备得到关键中间体(2r)-2-(反式-4-氨基环己基)-7-氯-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酰胺,再经1步反应得到伐美妥司他,本发明路线短,且通过拆分剂拆分得到单一构型r,优势明显。该方法具有原料易得,路线短,操作安全简单,产品纯度高,成本低等优点。
9、就本发明的具体工艺步骤而言,其创新之处在于:
10、在本发明中步骤1的反应中钌金属化合物催化剂为十二羰基三钌,尝试其他钌系复合物催化剂,如苯基亚甲基双(三环己基磷)二氯化钌(ii)、三(三苯基膦)羰基二氢钌、二(三苯基膦)环戊二烯基氯化钌,均无法达到十二羰基三钌与膦配体化合物共同催化的效果。
11、在本发明中步骤1反应中,十二羰基三钌与膦配体化合物共同催化的效果显著优于直接使用钌系复合物催化剂。
12、在本发明中步骤1反应中,膦配体化合物选自三(邻甲苯基)膦、三(对甲苯基)膦、三(间甲苯基)膦、三(3-甲氧基苯基)膦、三(2-甲氧基苯基)膦、三(3-甲氧基苯基)膦、5-二叔丁基膦-1',3',5'-三苯基-1'h-[1,4']二吡唑,优选三(邻甲苯基)膦、5-二叔丁基膦-1',3',5'-三苯基-1'h-[1,4']二吡唑,特别优选三(邻甲苯基)膦。
13、在本发明中步骤1的反应必须在非质子溶剂中进行才能最大限度降低副产物产生,另外考虑到反应温度和到物料的溶解特性,溶剂选自高沸点溶剂甲苯、二甲苯、二氧六环、dmf、dmso、n-甲基吡咯烷酮,优选为甲苯,从而调控反应温度和物料溶解性。
14、在本发明中步骤1的反应中,n-(反式-4-乙炔基环己基)氨基甲酸叔丁酯过量投料,可最大限度避免副产物产生,且过量物料可通过后处理萃取除去,但过量太多会增加物料成本。经研究摸索,优选了投料比例,控制中5-氯-n-((4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基)-3,4-二羟基-2-甲基苯甲酰胺和n-(反式-4-乙炔基环己基)氨基甲酸叔丁酯摩尔投料比为1:1-1:1.5,优选1:1.1-1:1.3,特别优选1:1.2。
15、在本发明中步骤1的反应中,辽金属化合物催化剂价格昂贵,其活性较高,优化其用量可大大降低生产成本,经研究摸索,优化5-氯-n-((4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基)-3,4-二羟基-2-甲基苯甲酰胺和辽金属化合物催化剂摩尔投料比为1:0.05-1:0.2,优选1:0.08-1:0.15,特别优选1:0.1,可保证反应完全,且用量最低。而膦配体化合物催化剂与辽金属化合物催化剂按摩尔比1:3配合使用形成复合物才能起到最佳催化效果,因此5-氯-n-((4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基)-3,4-二羟基-2-甲基苯甲酰胺和膦配体化合物催化剂摩尔投料比为1:0.2-1:0.4,优选1:0.25-1:0.35,特别优选1:0.3。
16、在本发明中步骤1的反应中,较高反应温度和较长反应时间均会导致副产物产生,经研究摸索,优化反应的条件包括:c-o交叉偶联反应阶段温度为80-100℃,优选在90-100℃搅拌反应5-7小时。脱保护反应阶段温度为20-30℃搅拌反应1-2小时,可最大限度减少副产物产生。
17、在本发明中步骤2中,(2rs)-7-氯-2-(反式-4-氨基环己基)-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酰胺拆分成单一r构型的方式为手性酸拆分或手性柱拆分,其中手性酸拆分收率高,且适宜产业化,作为优选。
18、在本发明中步骤2中,(2rs)-7-氯-2-(反式-4-氨基环己基)-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酰胺的手性酸拆分剂选择d-酒石酸、d-樟脑酸、d-二苯甲酰酒石酸、d-扁桃酸,其中d-酒石酸和d-樟脑酸拆分的光学纯度较高,作为优选,而d-酒石酸收率更高作为特别优选。
19、在本发明中步骤2的制备方法中,手性酸拆分剂过量导致s构型析出,影响光学纯度,手性酸拆分剂过少将导致收率偏低,经研究摸索确认(2rs)-7-氯-2-(反式-4-氨基环己基)-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酰胺与手性酸拆分剂的摩尔比投料比为1:0.4-1:0.6,优选1:0.45-1:0.55,特别优选1:0.5,即可保证光学纯度,同时可保证收率。
20、在本发明中步骤2的制备方法中,(2rs)-7-氯-2-(反式-4-氨基环己基)-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酰胺在醇类溶剂中通过手性酸拆分,醇类溶剂选自甲醇、无水乙醇、异丙醇,其中无水乙醇收率最高,且目的产物光学纯度高,作为优选。
21、在本发明中步骤2的制备方法中,所述步骤2中(2rs)-7-氯-2-(反式-4-氨基环己基)-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酰胺和手性酸在醇类溶剂中回流成盐溶解后,需先静置自然冷却至45-60℃,优选50-55℃,加入r构型目的产物的手性酸盐晶种,再继续静置自然冷却析晶,晶种加入对于保证目的产物光学纯度至关重要。
22、因此,本发明通过伐美妥司他中间体(2r)-7-氯-2-(反式-4-氨基环己基)-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酰胺新的合成路线设计,合成方法的优化,找到了一条路线更短、成本更低、更具产业化潜力的伐美妥司他的合成方法,并可从其中获益。
23、以下将结合实施例1-5以及对比例进一步说明伐美妥司他中间体(2r)-2-(反式-4-氨基环己基)-7-氯-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酰胺新的合成方法及由该方法的优势。该中间体(2r)-2-(反式-4-氨基环己基)-7-氯-n-[(4,6-二甲基-2-氧代-1,2-二氢吡啶-3-基)甲基]-2,4-二甲基-1,3-苯并二氧杂环戊烯-5-甲酰胺参考对比文件专利cn201680044712中实施例1的步骤1-6可制备得到伐美妥司他。
- 还没有人留言评论。精彩留言会获得点赞!