一种杂环化合物及其制备方法和应用与流程
本发明涉及药物,尤其涉及一种杂环化合物及其制备方法和应用。
背景技术:
1、癌症的特征是不定期的细胞增殖,抗细胞凋亡,永生复制,诱导血管生成,以及从身体的一个部位扩散到身体其他部位的潜力。在癌症中,基因突变导致细胞信号通路的改变,导致细胞代谢,细胞分化和肿瘤微环境的重组。cdks(ec2.7.11.22)是与细胞周期,转录,翻译和凋亡相关的丝氨酸/苏氨酸蛋白激酶。cdk的失调与肿瘤发生发展有关。(int.j.biol.macromol.2022,218,394-408)然而,cdk通过与细胞周期蛋白结合而活化。到目前为止,已经确定了至少20个cdk和30个细胞周期蛋白。其中cdk1,cdk2,cdk4和cdk6控制细胞周期中相的转变,而cdks 7-11在转录中发挥作用。在癌症,神经变性和肥胖症等疾病的进展期间,由于发生基因中的遗传突变和表观遗传突变而导致cdk的过表达或过度活化。这些发现表明通过抑制蛋白激酶活性是治疗这些疾病的一种新策略,换言之cdks可以作为治疗干预的靶点。
2、高度同源的cdk4和cdk6在细胞周期从g1期到s期的转变中起重要作用。在g1期的早期,细胞周期蛋白d与cdk4/6结合形成活化的cdk4/6-cyclin d复合物,进一步磷酸化视网膜母细胞瘤(rb)。随后,由于rb的磷酸化,转录因子e2f被释放;然后,加速从g1期到s期的转变,最终促进细胞周期和增殖。因此,癌基因cdk4/6可通过促进从g1期到s期的进展而引起不受控制的细胞增殖。(actapharm.sin.b 2021,11(1),30-54)cdk4/6的选择性抑制在癌症治疗中表现出有效的功效和低毒性。
3、目前,选择性cdk4/6抑制剂如palbociclib,ribociclib和abemaciclib已被美国食品和药物管理局(fda)批准用于治疗乳腺癌。(am.j.cancer res.2021,11(5),1913-1935)这些抑制剂可以分为两类,palbociclib和ribociclib属于一类,具有相似的结构,功效和毒性。用palbociclib或ribociclib连续治疗21天后,患者需要休息7天。abemaciclib是另一类,对cdk9表现出有效的抑制活性,但palbociclib和ribociclib对cdk9没有显示有效的抑制作用。患者可以连续给予abemaciclib,无需间歇给药。
4、选择性cdk4/6抑制剂在乳腺癌治疗中发挥了主导作用,2021年palbociclib,ribociclib和abemaciclib的总销售额超过70亿美元。然而,palbociclib和ribociclib可导致中性粒细胞减少症的不良事件,abemaciclib不仅抑制cdk4/6,还抑制cdk9。
技术实现思路
1、为了解决现有技术的问题,本发明实施例提供了一种杂环化合物及其制备方法和应用。所述技术方案如下:
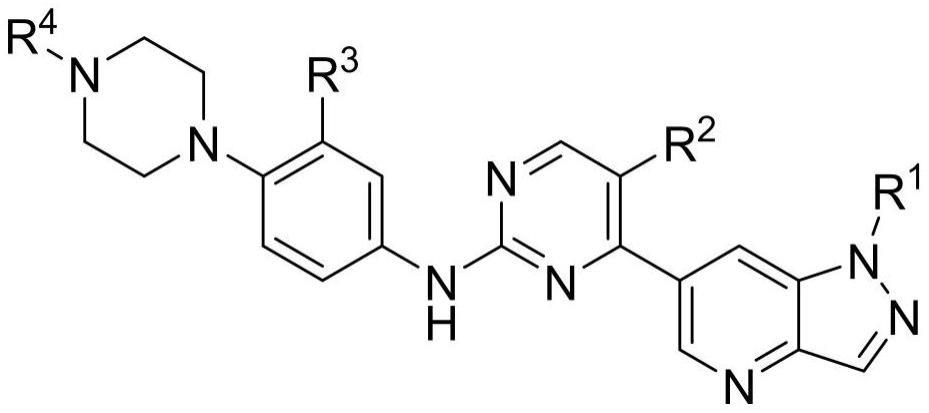
2、第一方面,提供一种杂环化合物及其药学上可接受盐,所述杂环化合物具有以下结构:
3、
4、其中,所述r1包括异丙基、乙基、环戊基、环丙基;r2包括甲基、氟;r3包括异丙基、氢、氟;r4包括异丙基、甲基、
5、进一步的,所述r1为异丙基,r2为甲基,r3为氢,r4为异丙基;或
6、所述r1为异丙基,r2为氟,r3为氢,r4为异丙基;或
7、所述r1为异丙基,r2为氟,r3为氢,r4为氢;或
8、所述r1为异丙基,r2为氟,r3为氢,r4为或
9、所述r1为异丙基,r2为氟,r3为氢,r4为或
10、所述r1为异丙基,r2为氟,r3为氢,r4为或
11、所述r1为异丙基,r2为氟,r3为氢,r4为或
12、所述r1为异丙基,r2为氟,r3为氢,r4为或
13、所述r1为异丙基,r2为氟,r3为氢,r4为甲基;或
14、所述r1为异丙基,r2为氟,r3为氟,r4为甲基;或
15、所述r1为异丙基,r2为氟,r3为氟,r4为异丙基;或
16、所述r1为异丙基,r2为氟,r3为氟,r4为氢;或
17、所述r1为乙基,r2为氟,r3为氢,r4为异丙基;或
18、所述r1为乙基,r2为氟,r3为氟,r4为异丙基;或
19、所述r1为环戊基,r2为氟,r3为氢,r4为异丙基;或
20、所述r1为环戊基,r2为氟,r3为氟,r4为异丙基;或
21、所述r1为环丙基,r2为氟,r3为氢,r4为异丙基;或
22、所述r1为环丙基,r2为氟,r3为氟,r4为异丙基。
23、第二方面,提供一种杂环化合物及其药学上可接受盐的制备方法,包括:
24、取化合物1在fe/nh4cl、meoh/h2o条件下80℃反应,得到化合物2;化合物2与乙酸酐的酰胺缩合反应得到化合物3;化合物3经过环化反应生成化合物4;然后将化合物4取下乙酰基,得到化合物5;化合物5在碳酸铯条件下与相应的卤代物反应得到化合物6a-6c;化合物6a-6c经硼酸取代反应得到化合物7a-7c,然后通过suzuki偶联反应得到化合物8a-8d;
25、
26、化合物9a与化合物10a在碳酸钾存在下反应得到化合物11a;化合物11a去除boc保护基团,得到化合物12a;化合物12a分别与相应的化合物8a-8d通过buchwald偶联反应,生成最终产物13a、13b、13m和13o;
27、
28、进一步的,取化合8b分别与化合物12d-12f进行buchwald偶联反应和de-boc反应,得到化合物13c、13i和13j;将化合物13c与相应的羧酸衍生物进行酰胺缩合反应,得到化合物13d-13h;
29、
30、进一步的,取化合物9b分别与化合物10a和10b进行c-n偶联反应,得到化合物11b和11c;化合物11b和11c在钯和h2的条件下被还原为化合物12b和12c;将化合物12b和12c分别与相应的化合物8b-8d进行buchwald偶联反应和de-boc保护基团反应,生成目标化合物13k、13l、13n和13p;
31、
32、进一步的,取化合物5与环丙基硼酸进行c-n偶联反应,得到化合物6d;合物6d经过硼酸盐取代反应得到化合物7d;化合物7d再经suzuki偶联反应得到中间体8e;化合物8与化合物12a和12b分别进行buchwald偶联反应,生成化合物13q和13r;
33、
34、第三方面,提供一种药物组合物,所述药物组合物包括上述第一方面所述的杂环化合物及其药学上可接受盐。
35、第四方面,提供一种如上述第一方面所述杂环化合物及其药学上可接受盐在制备抗肿瘤药物中的应用。
36、本发明实施例提供的技术方案带来的有益效果是:本发明提供一种杂环化合物13a-13h。化合物13a-13r对肿瘤细胞mda-mb-231和mcf-7都表现出很好的抑制活性。
- 还没有人留言评论。精彩留言会获得点赞!