聚羟基脂肪酸酯的提取方法与流程
1.本发明涉及生物工程技术及生物化工技术领域,具体涉及一种聚羟基脂肪酸酯(pha)的高纯度提取方法,包括物理、酶解、化学相结合的集成式提取工艺。
背景技术:
2.聚羟基脂肪酸酯是由很多细菌合成的一种胞内聚酯,在生物体内主要是作为碳源和能源的贮藏性物质而存在,它具有类似于合成塑料的物化特性及合成塑料所不具备的生物可降解性、生物相容性,所以pha属于环境友好型材料。
3.预计到2030年,塑料的年消耗量将达到7亿多吨。塑料制品的大规模应用,在带给人们便利的同时,也带来了严重的环境污染问题,pha则可以在自然化环境中降解为水和二氧化碳。因此开发一种低成本、高效率、高纯度提取pha的方法,使pha能够更好的进行商业化量产,有助于解决当前世界所面临的塑料污染问题。pha作为细菌内含物,胞内成分相当复杂,提取难度相当大,在pha发展的历程中,研究者们对提取做了大量的研究。目前提取pha的方法主要分为两大类型,一种是有机提取法,另外一种是水相提取法。
4.有机溶剂法如卤化物萃取溶剂如氯仿、二氯甲烷等的缺点是溶剂回收困难,生产环境危险,提取成本高昂;水相提取法的主要方式有物理法、酶解法、化学法等方法。物理提取法主要是对细胞进行机械破碎。但该方法能耗大,放大难度大,破壁不均匀,且单独通过物理方式破壁需要进行多次处理;化学法主要为次氯酸钠-sds法,缺点是次氯酸钠刺激性强,且含有sds的废水处理难度大;酶法的缺点是酶的用量大,部分酶单价高所以导致成本很高。
5.水相提取法有如下专利文献。
6.专利文献cn109504715a公开了一种制备聚羟基脂肪酸酯(pha)的方法,包括发酵液固液分离后洗涤,然后破壁。该方法虽然提取工艺简单,但是依然需要加入阴离子表面活性剂,污水较难处理,且该方法在破壁过程中需要保持高速离心状态,这就导致该方法能耗较高。
7.专利文献cn1464063a的发明专利公开了一种可有效降低pha分离提取成本的方法,该方法先用物理法对细菌进行破壁,然后用碱液调ph至碱性,在碱性的预处理液中,加入阴离子表面活性剂和凝聚剂,分离提取处理液中的沉淀,然后洗涤干燥,得到成品。该方法反应条件温和,设备简单,但要加入的表面活性剂为sds,刺激性较强,废水处理复杂。
8.专利文献cn111393625b的发明溶菌酶结合sds下利用超声提取pha的方法,该方法采用的表面活性剂为sds,废水中同样含有sds,且回收率和纯度不够理想。且该方法需要进行多次超声处理,能耗较高。
9.专利文献cn111346580b的发明公开了一种高压均质结合超声提取聚羟基脂肪酸酯的方法和系统。该方法虽然避免了强酸、强碱和sds带来的不利影响,但是该方法采用的纯物理的破壁方式为在高温高压蒸煮的同时进行第一次超声,离心处理重悬后进行第二次超声,其主要缺点在于能耗较高,且需要较高温度处理,超声破碎不易放大应用。
10.专利文献cn1190674的发明专利公开了一种从细菌菌体内分离提取pha的方法,包括如下步骤:1)用含有表面活性剂的碱性溶液处理菌体;2)固液分离,分离出去大部分非pha成分;3)用碱性蛋白酶处理pha;4)分离提取pha颗粒;5)干燥得到pha产品。该方法反应条件温和,但是耗碱量大,表面活性剂添加量大,碱性蛋白酶用量大,成本比较大。
技术实现要素:
11.为解决上述技术问题,本技术提供了一种聚羟基脂肪酸酯(pha)的高纯度绿色环保的提取方法,通过优化破壁条件综合物理、酶解、化学等破壁方法进行高效破壁,实现pha的高回收率和高纯度提取。用该方法获得的多种pha产品回收率达94%以上,纯度达98%以上。
12.本发明提供了一种聚羟基脂肪酸酯的提取方法,包括:a)从发酵液中分离获得菌体,将菌体重悬于水中获得细胞液,加入表面活性剂,然后进行破壁裂解处理,获得裂解液,将裂解液固液分离,弃上清,得到沉淀;b)将步骤a)中的沉淀重悬于水中获得裂解后悬液,向裂解后悬液中加入酶进行酶解,酶解后固液分离,弃上清,得到沉淀;c)将b)中获得的沉淀重悬得到酶解后悬液,加入漂白消毒剂后进行固液分离,得到沉淀。
13.所述的酶为生物降解酶。所述的酶为复合酶制剂,所述的复合酶制剂包括溶菌酶、核酸降解酶、葡聚糖酶、甘露聚糖酶、蜗牛酶、蛋白酶。
14.所述的复合酶制剂按照质量份数包括:溶菌酶大于3份;核酸降解酶大于0.5份;葡聚糖酶大于1份;甘露聚糖酶大于2份;蜗牛酶大于0.5份,优选大于1份;蛋白酶大于1份。
15.优选的,所述的复合酶制剂按照质量份数包括:溶菌酶:3-5份;核酸降解酶:0.5-2份;葡聚糖酶:1-3份;甘露聚糖酶:2-4份;蜗牛酶:0.5-2份,优选1-2份;蛋白酶:1-3份。
16.在本发明的一个具体实施方式中,所述的酶为复合酶制剂,所述的复合酶制剂包括四份溶菌酶、一份核酸降解酶、两份葡聚糖酶、三份甘露聚糖酶、一份蜗牛酶、两份蛋白酶。
17.所述的复合酶制剂通过将各种酶制剂粉末按照相应的比例称量并在混合器内混匀。
18.所述的酶按照质量百分比多于0.01%(优选0.01-2%,进一步优选0.01-0.08%,更进一步优选0.03%-0.05%)加入裂解后悬液中。例如按照质量百分比0.01%、0.02%、0.03%、0.04%、0.05%、0.06%、0.07%、0.08%、0.1%、0.2%、0.3%、0.4%、0.5%、1.0%、1.2%、1.5%、2.0%加入裂解后悬液中。
19.所述酶解的温度为40-60℃,例如40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60℃。
20.所述酶解的时间为10-200min,优选100-200min,例如10、20、50、100、110、120、130、140、150、160、170、180、190、200min。
21.所述的表面活性剂为环保型表面活性剂,优选的,所述的表面活性剂选自脂肪醇聚氧乙烯醚、脂肪醇聚氧乙烯醚硫酸盐、脂肪醇聚氧乙烯醚羧酸盐、α-烯基磺酸盐中的一种或两种以上的组合。
22.所述的表面活性剂的加入量为菌液干重的0.5%-5.0%,例如0.5%、1.0%、1.5%、2.0%、2.5%、3.0%、3.5%、4.0%、4.5%、5.0%。
23.所述的漂白消毒剂为饮用水漂白消毒剂,优选的,所述的漂白消毒剂选自过氧化丙酮、二氯异氰尿酸钠、二氧化氯中的一种或两种以上的组合。
24.所述的漂白消毒剂的加入量为酶解料液干重的0.01-1.5%,例如0.01、0.05、0.1、0.15、0.2、0.25、0.3、0.35、0.4、0.45、0.5、0.55、0.6、0.65、0.7、0.75、0.8、0.85、0.9、0.95、1、1.1、1.2、1.3、1.4、1.5%。
25.步骤a)中获得的细胞液中菌体干重为50-300g/l,优选100-280g/l;例如100、120、140、160、180、200、220、240、260、280g/l。
26.步骤b)中获得的裂解后悬液中水的加入量为步骤a)中弃去上清的体积。
27.步骤c中获得的酶解后悬液中水的加入量为步骤b)中弃去上清的体积。
28.所述的破壁裂解为一次或多次。例如1、2、3、4、5、6、7、8、9、10及以上次。
29.所述的破壁裂解选自高压均质、超声、物理研磨、高速珠磨法、温和碱处理法中的一种或两种以上的组合。
30.所述的破壁裂解时间为10-200min,优选100-200min,例如10、20、50、100、110、120、130、140、150、160、170、180、190、200min。
31.所述的高压均质中均质压力为0.5-1.2mpa;优选0.5-1.0mpa,例如0.5、0.6、0.7、0.8、0.9、1、1.1、1.2mpa。
32.所述的超声中功率为500-700w/m3;例如500、550、600、650、700w/m3。
33.所述的高速珠磨法中搅拌速度为650-1200r/min,优选为800-1200r/min,例如650、700、800、900、1000、1100、1200r/min。
34.所述的高速珠磨法中珠粒直径为0.05-0.2mm,优选0.05-0.1mm,例如0.05、0.1、0.15、0.2mm。
35.所述的温和碱处理法中ph值为8.5-11,优选9.5-11,例如8.5、9、9.5、10、10.5、11。
36.所述的温和碱处理法中温度为40-65℃,优选55-65℃,例如40、45、50、55、60、65℃。
37.所述的温和碱处理法中处理时间为50-70min,例如50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70min。
38.从发酵液中分离获得菌体包括将发酵液在7000-9000rpm(例如7000、7500、8000、8500、9000rpm)离心5-15min(例如5、8、10、12、15min),收集沉淀,即为菌体。
39.所述的加入酶或漂白消毒剂之后还包括离心、洗涤的步骤,优选重复离心、洗涤至少1、2、3、4、5、6、7、8、9、10及以上次。优选1-10次,进一步优选1-7次。
40.所述的提取方法还包括步骤d)将获得的沉淀一次或多次加水洗涤以及一次或多次(例如1、2、3、4、5、6、7、8、9、10或以上次)固液分离,弃上清,取沉淀干燥。
41.所述的固液分离为离心、絮凝分离或板框过滤。例如可以采用碟片离心机分离,絮凝分离,板框过滤分离,卧式螺旋离心分离、管式离心机分离,杯式离心机分离等等。
42.所述的干燥选自冷冻干燥、喷雾干燥、流化床干燥、转鼓干燥或沸腾床干燥中的一种或两种以上的组合。
43.所述的菌体选自嗜盐菌、兽气单胞菌、卡巴耶罗氏菌、大肠杆菌、酵母菌、乳酸菌、木质素降解细菌或巨大产碱杆菌dsm;其中,所述的嗜盐菌为盐单胞菌。
44.在本发明的一个具体实施方式中,所述的提取方法包括以下步骤:a)从发酵液中分离获得菌体,将菌体重悬于水中至干重50-300g/l获得细胞液,按照细胞干重的0.5-5.0%加入环保型表面活性剂;细胞干重优选为100-280g/l。
45.b)将该重悬液进行破壁裂解处理10-200min,获得破壁裂解液,破壁裂解方法可以为高压均质处理压力为0.5-1.2mpa,或者,高速珠磨的转速为650-1200r/min,粒径为0.05-0.2mm,或者,温和碱处理温度40-65℃,ph8.5-11处理获得破壁裂解液;优选的高压均质处理压力为0.5-1.0mpa,高速珠磨的转速为800-1200r/min,粒径为0.05-0.1mm,温和碱处理温度55-65℃,ph9.5-11。
46.c)离心、洗涤1-10次,优选的洗涤1-3次。
47.d)向洗涤后的破壁裂解液中按照质量百分比大于0.01%加入包括溶菌酶,核酸降解酶,葡聚糖酶、甘露聚糖酶、蜗牛酶和蛋白酶的复合酶制剂获得酶解液,所述酶解的温度为40-60℃,所述酶解的时间为10-200min;优选的按照质量百分比0.03%-0.08%向洗涤后的破壁裂解液中加入复合酶制剂。
48.e)离心、洗涤1-10次,优选的洗涤1-3次。
49.j)向上述酶解液按照料液干重的0.01-1.5%加入漂白消毒剂,优选按照料液干重的0.15%向酶解液中加入漂白消毒剂。
50.k)漂白后进行一次或多次的加水洗涤以及一次或多次的固液分离,获得的沉淀经干燥后即为聚羟基脂肪酸酯。
51.本发明还提供了一种复合酶制剂,所述的复合酶制剂包括溶菌酶、核酸降解酶、葡聚糖酶、甘露聚糖酶、蜗牛酶、蛋白酶。
52.所述的复合酶制剂按照质量份数包括:溶菌酶大于3份;核酸降解酶大于0.5份;葡聚糖酶大于1份;甘露聚糖酶大于2份;蜗牛酶大于0.5份,优选大于1份;
蛋白酶大于1份。
53.优选的,所述的复合酶制剂按照质量份数包括:溶菌酶:3-5份;核酸降解酶:0.5-2份;葡聚糖酶:1-3份;甘露聚糖酶:2-4份;蜗牛酶:0.5-2份,优选1-2份;蛋白酶:1-3份。
54.在本发明的一个具体实施方式中,所述的复合酶制剂包括四份溶菌酶、一份核酸降解酶、两份葡聚糖酶、三份甘露聚糖酶、一份蜗牛酶、两份蛋白酶。
55.本发明还提供了一种上述提取方法获得的聚羟基脂肪酸酯(pha)。
56.本发明还提供了一种上述提取方法获得的聚羟基脂肪酸酯(pha)在制备生物可降解材料中的应用。
57.溶菌酶,购自东恒华道生物技术有限公司,货号fdg-2270;核酸降解酶,购自湖北安琪酵母有限公司货号60675-83-4;蛋白酶:购自诺维信中国有限公司,货号:nspb0015;甘露聚糖酶、葡聚糖酶:购自广西庞博生物技术有限公司,货号分别为:37288-54-3和9025-70-1;蜗牛酶,购自西安奥肽生物科技有限公司,货号at-21122212。
58.有益效果:本方法采用了将多种破壁方式相结合的方法,使得细菌破壁成本大幅下降。本方法采用的破壁优于现有的技术特点在于采用了高菌体干重的重悬液(现有技术菌体干重为50-100g/l,本发明采用的菌体干重为100-280g/l),且由于后续还有其他破壁工艺,所以物理方式无需进行多次处理,一次即可,降低了现有技术能耗高的问题,且加入的表面活性剂为环保型表面活性剂,避免了现有技术加入sds刺激性强、导入污水较难处理的问题。
59.通过试验验证,采用本方法,即经过第一步物理方法后再采用酶解法,所需酶的用量将大大低于同类现有技术,但却能超过单一采用物理破碎或酶解法的提取效果,回收率和纯度都有显著提高,故本方法中酶解破壁以及整体的成本均得到了大幅度的降低。
60.本方法没有加入任何有机溶剂,成本低、污染小,相对其他水处理法,获得的产品纯度、回收率更高。
61.本方法采用的漂白剂均为饮用水消毒漂白剂。满足下游市场更全面的应用,适合产业化生产,更重要的是提取过程中产生的洗涤废水少,减轻固液分离的压力并减少化工试剂的使用(如强酸、强碱及有机试剂),实现绿色生产。
具体实施方式
62.在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
63.实施例中使用的多株产pha菌株,由本研究室筛选、改造及保藏,在摇瓶水平阶段细胞中的pha含量占细胞干重的80%左右。
64.实施例中提到的回收率的计算公式:回收率=c2w2/c1w1*100%c1:提取前细胞干物质含量w1:提取前pha纯度c2:提取后物料干物质含量w2:提取后pha纯度实施例中纯度的检测方法为:1. 标准曲线的制备1.1称样:称量5个标样作标准曲线,每个标样之间的质量间隔2 mg 左右;样品称量3个(称样量相近,作为平行实验)。
65.1.2 标准品的种类:1)phb(3-羟基丁酸,称量范围:18 mg-28 mg);2)γ-丁内酯(称量范围:2 ul-10 ul);3)hhx甲酯(3-羟基己酸甲酯,称量范围:10 mg-30 mg);4)phbv(聚3-羟基戊酸,称量范围:12 mg-30 mg)。
66.1.3 样品的种类:1)phb (包含3-羟基丁酸);2)p34hb (包含3-羟基丁酸和丁内酯);3)phbhhx (包含3-羟基丁酸和3-羟基己酸);4)phbv (包含3-羟基丁酸和3-羟基戊酸)。
67.2.酯化液的配制:取500ml无水甲醇(色谱纯),放入大烧杯中,缓慢加入15 ml浓硫酸,再加入0.5 g苯甲酸作为标准物质,转移到棕色瓶中放置过夜混匀。(注意事项:酯化液的配制要在通风橱中进行,酯化液提前24小时配好,用之前摇匀)3. 样品的制备:称取30-40 mg冷冻干燥后的菌体粉末放置于酯化管中。称取约10-20 mg pha标样于酯化管中作为参照,分别加入2 ml三氯甲烷与2 ml步骤1.1中配制的酯化液(注意:枪头用之前润洗),加盖密封后放入酯化仪,于100
º
c反应4 h。结束后,待酯化体系冷却至室温,加入1 ml去离子水,用涡旋振荡器充分振荡(转速1800,2分钟),静置分层1小时左右。使用注射器取下层有机相用于气相色谱分析。
68.4. 样品的分析: pha含量测定利用岛津公司gc-2014型气相色谱仪进行。仪器配置为:hp-5型色谱毛细管柱,氢火焰离子化检测器(flame ionization detector, fid),spl分流进样口;载气为高纯氮气,燃气为氢气,助燃气为空气;aoc-20s型自动进样器进样,以丙酮为洗涤液,在每次进样前洗涤3次,再用待测样品润洗。gc分析程序如下:检测器温度为250
º
c,进样口温度为240
º
c,色谱柱起始温度设为80
º
c,维持时间是1.5分钟,程序总时间一共为8分钟。样品测试结束后,通过内标归一法进行计算。
69.5. 气相色谱仪检测:1)样品准备:酯化液加水振荡,静置待分层(》1 h),用1 ml注射器吸取1 ml下层液体,注射到进样瓶。
70.2)瓶子安放:进样瓶专放座有顺序,从左到右(座上有数字)。一般一号放氯仿(用于跑基线和空白对照),其余样品按顺序依次摆放。
71.3)进样准备:跑样前检查清洗瓶里面的丙酮和氯仿是否足够。
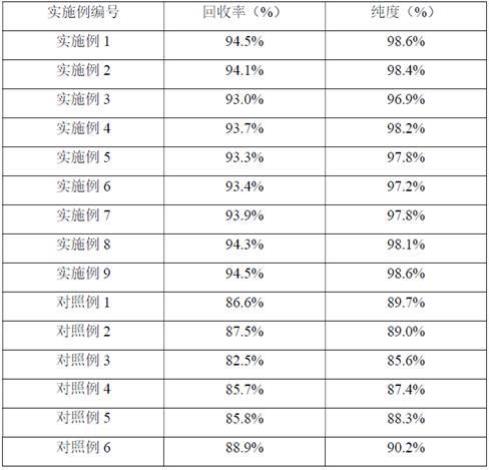
72.4)进样: 一般第一个样品放氯仿, 洗脱三次以平衡基线;第二个样品开始依次放待测样品,最后一个一般放标准品。为了减少误差,相同的样品要有至少三个平行样。
73.5)数据处理:每一次gc得到的数据主要看保留时间和对应峰面积,数据通过样品、内标准品(酯化液中的苯甲酸)和标准品进行分析。
74.实施例中复合酶制剂的制备方法:将各种酶制剂粉末按照相应的比例称量并在混合器内混匀。
75.实施例1(1)取嗜盐菌放罐发酵液200ml,进行8000rpm离心10min,弃去上清液,收集菌体沉淀。
76.(2)向步骤(1)中的菌体加入适量水,加入水的体积为原来菌液体积的一半,震荡混匀,获得100ml细胞液。
77.(3)向步骤(2)中的细胞液加入环保型表面活性剂脂肪醇聚乙烯醚硫酸盐(aes)母液(20%浓度)添加量0.75ml,搅拌处理20min。
78.(4)将步骤(3)中的获得的料液用高压均质机进行高压均质处理,均质压力为0.8mpa,均质后离心处理,弃去上清液,收集沉淀。
79.(5)将步骤(4)中收集的沉淀加水重悬,加入水的体积为弃去上清体积,震荡摇匀,获得裂解后悬液。
80.(6)向步骤(5)中的裂解后悬液加入0.03%(质量百分比)复合酶制剂(质量份数:四份溶菌酶、一份核酸降解酶、两份葡聚糖酶、三份甘露聚糖酶、一份蜗牛酶、两份蛋白酶)45℃处理120min。处理完成后进行离心处理,弃去上清液,收集沉淀。
81.(7)将步骤(6)中收集的沉淀加水重悬,加入水的体积为弃去上清体积,震荡摇匀,获得酶解后悬液。
82.(8)向步骤(7)中的酶解后悬液加入0.15g的二氧化氯,搅拌处理30min。处理完成后进行离心处理,弃去上清液,收集沉淀。
83.(9)将步骤(8)中获得的沉淀加水重悬,加入水的体积为弃去上清体积,搅拌处理30min后,离心处理,弃去上清液,收集沉淀。
84.(10)将步骤(9)得到的沉淀进行真空冷冻干燥。回收率和纯度如表1所示。分子量如表4所示。
85.实施例2本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是步骤(4)所述的物理破壁方法为超声破碎法,具体为将步骤(3)中的获得的料液用超声破碎法处理,超声功率为600w/m
³
,超声后离心处理,弃去上清液,收集沉淀。
86.收率和纯度如表1所示。
87.实施例3本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方
法进行聚羟基脂肪酸酯的分离,不同的是步骤(4)所述的物理破壁方法为高速珠磨法,具体为将步骤(3)中的获得的料液用高速珠磨法处理,搅拌速度为1200r/min,料液循环次数为1次,珠粒直径为0.1mm,然后离心处理,弃去上清液,收集沉淀。
88.收率和纯度如表1所示。
89.实施例4本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是步骤(4)中所述的物理破壁方法为温和碱处理法,具体为将步骤(3)中的获得的料液用温和碱处理法处理,ph值为10.0温度为65℃,处理时间60min,然后离心处理,弃去上清液,收集沉淀。
90.回收率和纯度如表1所示。
91.实施例5本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是步骤(3)中所述的环保型表面活性剂为脂肪醇聚氧乙烯醚(aeo),添加比例为细胞干重的0.5%,即0.75ml。回收率和纯度如表1所示。
92.实施例6本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是步骤(3)中所述的环保型表面活性剂为脂肪醇聚氧乙烯醚羧酸盐(aec),添加比例为细胞干重的0.5%,即0.75ml。回收率和纯度如表1所示。
93.实施例7本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是步骤(8)中二氧化氯替换为二氯异氰尿酸钠,添加量为0.15%,即0.15g。回收率和纯度如表1所示。
94.实施例8本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是步骤(8)中二氧化氯替换为过氧化丙酮,添加量为0.15ml。回收率和纯度如表1所示。
95.实施例9(1)取嗜盐菌放罐发酵液200ml,进行8000rpm离心10min,弃去上清液,收集菌体沉淀。
96.(2)向步骤(1)中的菌体加入适量水,加入水的体积为原来菌液体积的一半,震荡混匀,获得100ml细胞液。
97.(3)向步骤(2)中的细胞液加入环保型表面活性剂脂肪醇聚乙烯醚硫酸盐(aes)母液(20%浓度)添加量0.75ml,搅拌处理20min。
98.(4)将步骤(3)中的获得的料液用高压均质机进行高压均质处理,均质压力为0.8mpa,均质后离心处理,弃去上清液,收集沉淀。
99.(5)将步骤(4)中收集的沉淀加水重悬,加入水的体积为弃去上清体积,震荡摇匀,获得裂解后悬液。
100.(6)向步骤(5)中的裂解后悬液加入0.03%(质量百分比)复合酶制剂(质量份数:四份溶菌酶、一份核酸降解酶、两份葡聚糖酶、三份甘露聚糖酶、一份蜗牛酶、两份蛋白酶)45
℃处理120min。处理完成后进行离心处理,弃去上清液,收集沉淀。
101.(7)将步骤(6)中收集的沉淀加水重悬,加入水的体积为弃去上清体积,震荡摇匀,获得酶解后悬液。
102.(8)向步骤(7)中的酶解后悬液加入0.15g的二氯异腈脲酸钠,搅拌处理30min。处理完成后进行离心处理,弃去上清液,收集沉淀。
103.(9)将步骤(8)中获得的沉淀加水重悬,加入水的体积为弃去上清体积,搅拌处理30min后,离心处理,弃去上清液,收集沉淀。
104.(10)将步骤(9)得到的沉淀进行真空冷冻干燥。回收率、纯度如表1所示。
105.对照例1本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是不进行步骤(3)操作,即不加入任何表面活性剂,将步骤(2)获得的细胞液直接进行高压均质等后续处理。回收率和纯度如表1所示。
106.对照例2本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是不进行步骤(4)中部分操作,即不进行物理破壁方式,将步骤(3)获得的料液直接进行离心处理,弃去上清液,收集沉淀以及后续操作。回收率和纯度如表1所示。
107.对照例3本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是不进行步骤(6)中部分操作,即不进行酶解方式,将步骤(5)获得的裂解后悬液直接进行离心处理,弃去上清液,收集沉淀以及后续操作。回收率和纯度如表1所示。
108.对照例4本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是不进行步骤(8)中部分操作,即不进行漂白剂方式,将步骤(7)获得的酶解后悬液直接进行离心处理,弃去上清液,收集沉淀以及后续操作。回收率和纯度如表1所示。
109.对照例5本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是不进行步骤(6)中部分操作,即将步骤(5)获得的裂解后悬液直接离心处理,弃去上清液,收集沉淀,并且将步骤(4)重复操作3次。回收率和纯度如表1所示。
110.对照例6本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是不进行步骤(4)中部分操作,即将步骤(3)获得的料液直接进行离心处理,弃去上清液,收集沉淀,将步骤(6)中的复合酶制剂添加量提升至0.2%。回收率和纯度如表1所示。
111.表1
由表1可知采用本方法进行pha提取收率可以达到94%以上,纯度可以达到98%以上。而且对照例基本未能达到该水平。
112.对照例7本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是步骤(3)中所述的环保型表面活性剂为十二烷基硫酸钠sds,添加比例为细胞干重的0.5%,即0.75ml。回收率和纯度如表2所示。
113.表2由表2可知采用新型的环保表面活性剂的效果要优于sds。pha提取收率可以达到94%以上,纯度可以达到98%以上。
114.对照例8本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是步骤(6)复合酶制剂添加量为0.005%。回收率和纯
度如表3所示。
115.对照例9本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是步骤(6)复合酶制剂添加量为0.1%。回收率和纯度如表3所示。
116.对照例10本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是步骤(6)中复合酶制剂,添加量比例为1.2%。回收率和纯度如表3所示。
117.对照例11本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是步骤(6)中复合酶制剂,添加量比例为2.0%。回收率和纯度如表3所示。
118.表3由表3可知复合酶制剂的添加量在本发明申请范围内有着更好的效果,少于该添加量则pha纯度大幅度降低,高于该添加量也并未能取得更好的效果。
119.对照例12本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是步骤(8)中的药品替换为0.5%的双氧水。回收率、纯度、分子量如表4所示。
120.对照例13本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是步骤(8)中的药品替换为0.15%的次氯酸钠。回收率、纯度、分子量如表4所示。
121.表4由表4可知不同种类的漂白消毒剂对pha提取的分子量存在差较大。而本方法采用
的漂白消毒剂在防止pha分子量降低方面有较为明显的效果。
122.对照例14本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是步骤(6)中的复合酶制剂中不含有蜗牛酶,即配方调整为按照质量份数:四份溶菌酶、一份核酸降解酶、两份葡聚糖酶、三份甘露聚糖酶、两份蛋白酶。回收率、纯度如表5所示。
123.对照例15本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是步骤(6)中的复合酶制剂中不含有蛋白酶,即配方调整为按照质量份数:四份溶菌酶、一份核酸降解酶、两份葡聚糖酶、三份甘露聚糖酶、一份蜗牛酶。回收率、纯度如表5所示。
124.表5对照例16本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是步骤(6)中的复合酶制剂中蜗牛酶的量调整为0.5、2、5份,即配方调整为按照质量份数:四份溶菌酶、一份核酸降解酶、两份葡聚糖酶、三份甘露聚糖酶、蜗牛酶0.5份或2份或5份、两份蛋白酶。回收率、纯度如表6所示。
125.表6对照例17本实施例用于说明本发明提供的聚羟基脂肪酸酯的分离方法。按照实施例1的方法进行聚羟基脂肪酸酯的分离,不同的是步骤(6)中的复合酶制剂中蛋白酶的量调整为1份,即配方调整为按照质量份数:四份溶菌酶、一份核酸降解酶、两份葡聚糖酶、三份甘露聚糖酶、一份蜗牛酶、一份蛋白酶。回收率、纯度如表7所示。
126.表7
以上详细描述了本发明的优选实施方式,但是,本发明中的微生物并不限于上述实施方式中提到的嗜盐菌,在本发明的技术构思范围内,可以对本发明的微生物进行更换,更换为其他产pha的微生物,这些简单的更换均属于本发明的保护范围。
127.另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1