非奈利酮及其中间体的制备方法与流程
本发明涉及非奈利酮及其中间体的制备方法。
背景技术:
1、非奈利酮,化学名(4s)-4-(4-氰基-2-甲氧基苯基)-5-乙氧基-2,8-二甲基-1,4-二氢-1,6-萘啶-3-甲酰胺,作为盐皮质激素受体的非甾体拮抗剂,可用作预防和/或治疗心血管和肾脏疾病,例如心力衰竭和慢性肾病。
2、
3、chemmedchem,2012,7,1385报道了,从香草醛出发,10步反应制备非奈利酮,总收率仅为3.76%。
4、wo2008/104306也报到了非奈利酮的制备方法,但是因为总收率较低(只有5%)、许多中间色还需要谱纯化,且需要大量消耗溶剂,故不适合商业化生产。wo 2008/104306中,为拆分使用了专门合成的手性相(内部制备),其包含聚(n-甲基丙烯酰基-d-亮氨酸-二环丙基甲基酰胺)作为手性选择剂。据拜耳报道,还可以在容易商购获得的相上进行分离。其采用相chiralpak as-v,20μm的形式。所用洗脱液为甲醇/乙腈60∶40的混合物。在这种情况下,色谱分析可在常规色谱柱上进行,但优选使用本领域技术人员已知的技术,例如smb(模拟移动床;g.paredes,m.mazotti,journal of chromatography a,1142(2007):56-68)或varicol(computers and chemicalengineering 27(2003)1883-1901)。
5、cn112040318a报道了采用通式(iiia)或(iiib)的手性取代的酒石酸酯作为拆分剂进行外消旋体拆分,得到非奈利酮,拆分收率达到91.4%。但是该方法拆分后,残留的拆分剂较难去除。
6、因此,需要寻找工业上可行、总收率高、生产成本低和制得产品纯度高的非奈利酮的制备方法。
技术实现思路
1、本发明所要解决的技术问题是为了克服现有技术中非奈利酮的合成方法反应步骤长、总收率低、后处理步骤繁琐、制得的产品纯度低、拆分步骤繁琐、生产成本高、不适合于工业化生产等缺陷而提供了非奈利酮及其中间体的制备方法。本发明的制备方法,反应步骤短、反应总收率高、操作简单安全、后处理步骤简单、制得的产品纯度高、生产成本低、适合于工业化生产。
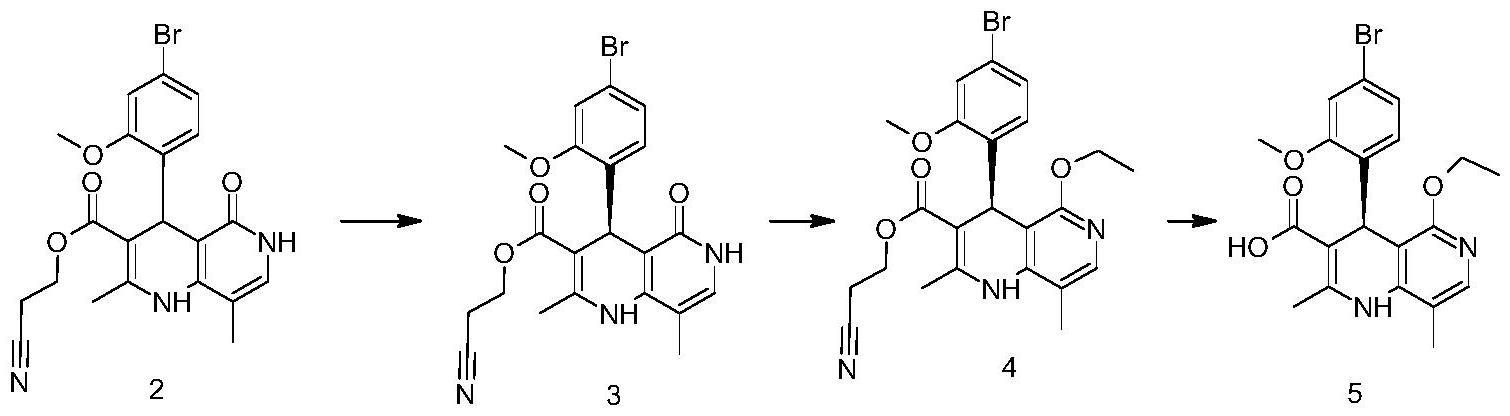
2、本发明提供了一种非奈利酮中间体3的制备方法,其包括以下步骤:有机溶剂中,将非奈利酮中间体2与d-酒石酸二苯酯进行拆分反应,得到拆分盐,然后与碱进行酸碱反应,得到所述的非奈利酮中间体3即可;
3、
4、本发明中,所述的非奈利酮中间体3的制备方法,优选采用以下反应条件:
5、非奈利酮中间体3的制备方法中,所述的有机溶剂优选卤代烃类溶剂;所述的卤代烃类溶剂优选二氯甲烷。
6、非奈利酮中间体3的制备方法中,所述的有机溶剂与所述的非奈利酮中间体2的体积质量比值优选1g/ml~50ml,进一步优选20g/ml~40ml,例如30ml。
7、非奈利酮中间体3的制备方法中,所述的d-酒石酸二苯酯与所述的非奈利酮中间体2的摩尔比值优选0.5~3.0,进一步优选0.8~1.5,例如1.1。
8、非奈利酮中间体3的制备方法中,所述的拆分反应的温度优选10℃~50℃,进一步优选20℃~40℃,例如30℃~40℃或者20℃~25℃。
9、非奈利酮中间体3的制备方法中,所述的拆分反应的优选1小时~24小时,进一步优选18小时~20小时。
10、非奈利酮中间体3的制备方法中,所述的碱可以为磷酸钠、碳酸钠和碳酸氢钠中的一种或多种。
11、非奈利酮中间体3的制备方法中,所述的酸碱反应的ph优选7-9。
12、非奈利酮中间体3的制备方法中,所述的酸碱反应的温度优选20℃~80℃,例如50℃~70℃。
13、非奈利酮中间体3的制备方法中,所述的酸碱反应的时间优选1小时~30小时,进一步优选5小时~15小时。
14、本发明所述的非奈利酮中间体3的制备方法,进一步包括非奈利酮中间体2的制备方法,其包括以下步骤:有机溶剂中,将非奈利酮中间体1与4-氨基-5-甲基-2-羟基吡啶进行合环反应,得到所述的非奈利酮中间体2即可;
15、
16、非奈利酮中间体2的制备方法可以采用本领域中该类合环反应的常规条件,本发明中优选以下反应条件:
17、非奈利酮中间体2的制备方法中,所述的有机溶剂优选醇类溶剂;所述的醇类溶剂优选2-丁醇(仲丁醇)和/或1-丁醇(正丁醇)。
18、非奈利酮中间体2的制备方法中,所述的有机溶剂与所述的非奈利酮中间体1的体积质量比值优选1ml/g~30ml/g;进一步优选1ml/g~5ml/g,例如2ml/g。
19、非奈利酮中间体2的制备方法中,所述的非奈利酮中间体1与所述的4-氨基-5-甲基-2-羟基吡啶的质量比值优选1.0~5.0,进一步优选2.0~3.0,例如2.6。
20、非奈利酮中间体2的制备方法中,所述的环化反应的温度优选80℃~150℃,进一步优选90℃~140℃,例如120℃。
21、非奈利酮中间体2的制备方法中,所述的环化反应的时间采用本领域常规检测方法(例如hplc、nmr或tlc)进行监测,一般以所述的非奈利酮中间体1消失时为反应的终点,优选1小时~24小时,进一步优选10小时~20小时,例如16小时。
22、非奈利酮中间体2的制备方法优选采用以下后处理步骤,反应结束后,降温、析晶、打浆得到所述的非奈利酮中间体2。所述的降温优选降至温度40℃~50℃。所述的析晶的温度优选0~10℃。所述的析晶的时间优选1小时~10小时,例如2小时~3小时。所述的打浆优选采用柠檬酸水溶液。所述的柠檬酸水溶液的浓度优选1%~10%,例如5%,所述的浓度是指柠檬酸的质量占柠檬酸水溶液总质量的百分比。
23、本发明所述的非奈利酮中间体3的制备方法,再进一步包括非奈利酮中间体1的制备方法,其包括以下步骤:有机溶剂中,酸和催化剂存在下,将4-溴-2-甲氧基苯甲醛与2-氰基乙酰乙酸乙酯进行缩合反应,得到所述的非奈利酮中间体1即可;
24、
25、非奈利酮中间体1的制备方法可以采用本领域中该类缩合反应的常规方法,本发明特别优选以下反应条件:
26、非奈利酮中间体1的制备方法中,所述的有机溶剂优选醇类溶剂;所述的醇类溶剂优选异丙醇。
27、非奈利酮中间体1的制备方法中,所述的有机溶剂与所述的4-溴-2-甲氧基苯甲醛的体积质量比值优选1.0ml/g~30.0ml/g,进一步优选2.0ml/g~10.0ml/g,例如3.3ml/g。
28、非奈利酮中间体1的制备方法中,所述的2-氰基乙酰乙酸乙酯与所述的4-溴-2-甲氧基苯甲醛的质量比值优选1.0~5.0,进一步优选1.0~2.0,例如1.0。
29、非奈利酮中间体1的制备方法中,所述的酸优选有机酸;所述的有机酸优选乙酸。
30、非奈利酮中间体1的制备方法中,所述的酸与所述的4-溴-2-甲氧基苯甲醛的摩尔比值优选0.01~1,进一步优选0.10~0.30,例如0.15。
31、非奈利酮中间体1的制备方法中,所述的催化剂优选哌啶。
32、非奈利酮中间体1的制备方法中,所述的催化剂与所述的4-溴-2-甲氧基苯甲醛的摩尔比值优选0.01~1,进一步优选0.10~0.30,例如0.15。
33、非奈利酮中间体1的制备方法中,所述的缩合反应的温度优选10℃~60℃,进一步优选20℃~50℃,例如30℃~40℃。
34、非奈利酮中间体1的制备方法中,所述的缩合反应的时间可以采用本领域中的常规检测方法(例如hplc、tlc或nmr)进行监测,一般以所述的4-溴-2-甲氧基苯甲醛消失时为反应的终点,本发明中优选1小时~24小时,例如3小时~4小时。
35、非奈利酮中间体1的制备方法优选包括以下后处理步骤:反应结束后,析晶、过滤、洗涤得到所述的非奈利酮中间体1。所述的析晶的温度优选10℃~20℃。所述的析晶的时间优选1小时~24小时,例如16小时。所述的洗涤优选采用醇类溶剂;所述的醇类溶剂优选甲醇。
36、本发明还提供了非奈利酮中间体4的制备方法,其包括以下步骤:按照上述方法制得非奈利酮中间体3之后,再在有机溶剂中,酸存在下,将非奈利酮中间体3与原甲酸三乙酯进行亲核取代反应,得到所述的非奈利酮中间体4即可;
37、
38、非奈利酮中间体4的制备方法可以采用本领域中该类亲核取代反应的常规条件,本发明中特别优选以下反应条件:
39、非奈利酮中间体4的制备方法中,所述的有机溶剂优选酰胺类溶剂;所述的酰胺类溶剂优选n,n-二甲基乙酰胺和/或n,n二甲基甲酰胺。
40、非奈利酮中间体4的制备方法中,所述的有机溶剂与所述的非奈利酮中间体3的体积质量比优选1ml/g~100ml/g,进一步优选2ml/g~10ml/g,例如5.7ml/g。
41、非奈利酮中间体4的制备方法中,所述的原甲酸三乙酯与所述的非奈利酮中间体3的质量比值优选1~5,进一步优选1.1~2.0,例如1.4。
42、非奈利酮中间体4的制备方法中,所述的酸优选无机酸,所述的无机酸优选浓硫酸。所述的浓硫酸可以为常规市售浓硫酸试剂。所述的浓硫酸的浓度可以为98%,所述的浓度是指硫酸的质量占浓硫酸水溶液总质量的百分比。
43、非奈利酮中间体4的制备方法中,所述的酸与所述的非奈利酮中间体3的摩尔比值优选0.10~0.50,进一步优选0.20~0.45,例如0.38。
44、非奈利酮中间体4的制备方法中,所述的亲核取代反应的温度优选100℃~150℃,进一步优选110℃~140℃,例如115℃~125℃。
45、非奈利酮中间体4的制备方法中,所述的亲核取代反应的时间采用本领域中的常规检测方法(例如tlc、hplc或nmr)进行监测,一般以所述的非奈利酮中间体3消失时为反应的终点,所述的亲核取代反应的时间优选1小时~10小时,例如2小时~3小时。
46、非奈利酮中间体4的制备方法优选包括以下后处理步骤,反应结束后,降温、加水、析晶,得到所述的非奈利酮中间体4即可。所述的降温可以降至50℃~60℃。所述的析晶优选加入晶种搅拌析晶;所述的析晶的温度优选0~10℃。所述的析晶的时间优选2小时~3小时。
47、本发明还提供了非奈利酮中间体5的合成方法,其包括以下步骤:按照上述方法制得非奈利酮中间体4之后,再在溶剂中,将非奈利酮中间体4进行水解反应,得到所述的非奈利酮中间体5即可;
48、
49、非奈利酮中间体5的制备方法可以采用本领域中该类水解反应的常规条件,本发明中特别优选以下反应条件:
50、非奈利酮中间体5的制备方法中,所述的有机溶剂优选醚类溶剂;所述的醚类溶剂优选四氢呋喃(thf)。
51、非奈利酮中间体5的制备方法中,所述的有机溶剂与所述的非奈利酮中间体4的体积质量比优选1ml/g~100ml/g,进一步优选2ml/g~10ml/g,例如5.3ml/g。
52、非奈利酮中间体5的制备方法中,所述的碱优选无机碱,所述的无机碱优选氢氧化钠。所述的氢氧化钠可以以水溶液的形式使用。所述的氢氧化钠水溶液的浓度可以为1%~50%,例如7.3%,所述的百分比是指氢氧化钠的质量占氢氧化钠水溶液总质量的百分比。
53、非奈利酮中间体5的制备方法中,所述的碱与所述的非奈利酮中间体4的摩尔比值优选1~5,进一步优选1.1~3.0,例如2.0。
54、非奈利酮中间体5的制备方法中,所述的水解反应的温度优选-10℃~20℃,进一步优选-5℃~10℃,例如-5℃~5℃。
55、非奈利酮中间体5的制备方法中,所述的水解反应的时间采用本领域中的常规检测方法(例如tlc、hplc或nmr)进行监测,一般以所述的非奈利酮中间体4消失时为反应的终点,所述的水解反应的时间优选1小时~10小时,例如4小时~5小时。
56、非奈利酮中间体5的制备方法优选包括以下后处理步骤,反应结束后,萃取、调节ph3左右、过滤、洗涤,得到所述的非奈利酮中间体5即可。所述的萃取优选采用甲苯。所述的调节ph可以采用盐酸;所述的盐酸的浓度可以为5%~15%,例如10%,所述的百分比是指氯化氢的质量占盐酸水溶液总质量的百分比。所述的洗涤优选依次采用水及甲苯洗涤;所述的洗涤的次数优选1~3次,例如2次。
57、本发明还提供了非奈利酮中间体6的制备方法,其包括以下步骤:按照上述方法制得非奈利酮中间体5之后,再在有机溶剂中,催化剂和缩合剂存在下,将非奈利酮中间体5与六甲基二氮硅烷进行缩合反应,得到所述的非奈利酮中间体6即可;
58、
59、所述的非奈利酮中间体6的制备方法可以采用本领域中该类缩合反应的常规方法和条件,本发明特别优选以下反应条件:
60、非奈利酮中间体6的制备方法中,所述的有机溶剂优选醚类溶剂;所述的醚类溶剂优选四氢呋喃(thf)。
61、非奈利酮中间体6的制备方法中,所述的有机溶剂与所述的非奈利酮中间体5的体积质量比优选1ml/g~30ml/g,进一步优选2ml/g~10ml/g,例如5.2ml/g。
62、非奈利酮中间体6的制备方法中,所述的催化剂优选4-二甲氨基吡啶(dmap)。
63、非奈利酮中间体6的制备方法中,所述的催化剂与所述的非奈利酮中间体5的摩尔比值优选0.01~2,进一步优选0.05~0.2,例如0.1。
64、非奈利酮中间体6的制备方法中,所述的缩合剂优选n,n′-羰基二咪唑(cdi)。
65、非奈利酮中间体6的制备方法中,所述的缩合剂与所述的非奈利酮中间体5的摩尔比值优选1.0~5.0,进一步优选1.1~2.0,例如1.4。
66、非奈利酮中间体6的制备方法中,所述的六甲基二硅氮烷与所述的非奈利酮中间体5的摩尔比值优选1.0~10.0,进一步优选2.0~6.0,例如4.4。
67、非奈利酮中间体6的制备方法中,所述的缩合反应的温度优选20℃~100℃,进一步优选65℃~75℃,例如70℃。
68、非奈利酮中间体6的制备方法中,所述的缩合反应的时间采用本领域中的常规检测方法(例如tlc、hplc或nmr)进行监测,一般以所述的非奈利酮中间体5消失时为反应的终点,所述的缩合反应的时间优选5小时~25小时,进一步优选10小时~20小时,例如16小时。
69、非奈利酮中间体6的制备方法优选包括以下后处理步骤,反应结束后,加四氢呋喃水溶液、回流、降温、过滤、洗涤,得到所述的非奈利酮中间体6即可。所述的四氢呋喃水溶液中四氢呋喃与水的体积比值优选0.5~5,例如1.4。所述的回流的温度优选70℃~80℃。所述的降温优选降至0℃左右。所述的降温的速率优选14℃/小时~27℃/小时。所述的洗涤优选依次采用四氢呋喃及水洗涤;所述的洗涤的次数优选1~3次,例如2次。
70、本发明还提供了非奈利酮的制备方法,其包括以下步骤:按照上述方法制得非奈利酮中间体6之后,再在有机溶剂中,催化剂存在下,将非奈利酮中间体6与氰化锌进行亲核取代反应,得到非奈利酮即可;
71、
72、所述的非奈利酮的制备方法可以采用本领域中该类亲核取代反应的常规方法和条件,本发明特别优选以下反应条件:
73、非奈利酮的制备方法中,所述的有机溶剂优选酰胺类溶剂;所述的酰胺类溶剂优选n,n-二甲基甲酰胺(dmf)。
74、非奈利酮的制备方法中,所述的有机溶剂与所述的非奈利酮中间体6得体积质量比值优选1ml/g~30ml/g,进一步优选2ml/g~20ml/g,例如10ml/g。
75、非奈利酮的制备方法中,所述的氰化锌与所述的非奈利酮中间体6的摩尔比值优选0.5~3,进一步优选1.0~2.0,例如1.5。
76、非奈利酮的制备方法中,所述的催化剂优选1,1'-双(二苯基膦)二茂铁和/或pd2(dba)3,四三苯基磷钯。
77、非奈利酮的制备方法中,所述的催化剂与所述的非奈利酮中间体6的摩尔比值优选0.001~1.0,进一步优选0.05~0.2,例如0.10。
78、非奈利酮的制备方法中,所述的亲核取代反应的温度优选60℃~150℃,进一步优选90℃~140℃,例如100℃。
79、非奈利酮的制备方法中,所述的亲核取代反应的时间可以采用本领域中的常规检测方法(例如hplc、tlc或nmr)进行监测,一般以所述的非奈利酮中间体6消失时为反应的终点,本发明中优选1小时~30小时,进一步优选5小时~25小时,例如15小时~20小时。
80、非奈利酮的制备方法优选包括以下后处理步骤:反应结束后,降温、萃取、洗涤、干燥得到非奈利酮粗品。非奈利酮粗品优选经过重结晶得到非奈利酮。所述的重结晶采用的溶剂优选醇类溶剂;所述的醇类溶剂优选乙醇。
81、本发明所述的非奈利酮的制备方法,优选采用以下合成路线:
82、
83、本发明还提供了非奈利酮中间体1的制备方法,其包括以下步骤:有机溶剂中,酸和催化剂存在下,将4-溴-2-甲氧基苯甲醛与2-氰基乙酰乙酸乙酯进行缩合反应,得到所述的非奈利酮中间体1即可;
84、
85、其中,各反应条件均同前所述。
86、本发明还提供了非奈利酮中间体2的制备方法,其包括以下步骤:有机溶剂中,将非奈利酮中间体1与4-氨基-5-甲基-2-羟基吡啶进行合环反应,得到所述的非奈利酮中间体2即可;
87、
88、其中,各反应条件均同前所述。
89、本发明还提供了非奈利酮中间体4的制备方法,其包括以下步骤:在有机溶剂中,酸存在下,将非奈利酮中间体3与原甲酸三乙酯进行亲核取代反应,得到非奈利酮中间体4即可;
90、
91、其中,各反应条件均同前所述。
92、本发明还提供了非奈利酮中间体5的制备方法,其包括以下步骤:在溶剂中,将非奈利酮中间体4进行水解反应,得到所述的非奈利酮中间体5即可;
93、
94、其中,各反应条件均同前所述。
95、本发明还提供了非奈利酮中间体6的制备方法,其包括以下步骤:在有机溶剂中,催化剂和缩合剂存在下,将非奈利酮中间体5与六甲基二氮硅烷进行缩合反应,得到所述的非奈利酮中间体6即可;
96、
97、其中,各反应条件均同前所述。
98、本发明还提供了非奈利酮的制备方法,其包括以下步骤:在有机溶剂中,催化剂存在下,将非奈利酮中间体6与氰化锌进行亲核取代反应,得到非奈利酮即可;
99、
100、其中,各反应条件均同前所述。
101、本发明中所述原料或试剂除特别说明之外,均市售可得。
102、本发明中,所述的室温是指环境温度,为10℃~35℃。
103、本发明的积极进步效果在于:本发明的制备方法,反应步骤短、反应总收率高、操作简单安全、后处理步骤简单、制得的产品纯度高、生产成本低、适合于工业化生产。
- 还没有人留言评论。精彩留言会获得点赞!