bufogargarizinA的制备方法
本发明涉及有机化学合成领域,特别是涉及一种bufogargarizin a的制备方法。
背景技术:
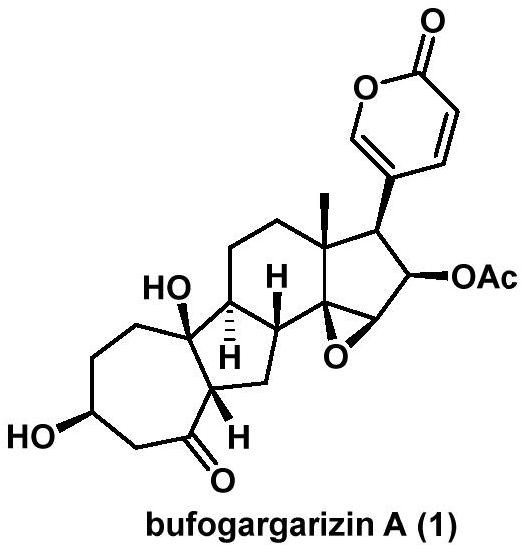
1、天然产物bufogargarizin a是由暨南大学的叶文才教授及其合作学者于2010年在具有抗炎症与抗癌活性的中华大蟾蜍的毒液中提取出来的一种活性甾体类化合物,其化学结构如下所示:
2、
3、在临床中,蟾蜍中的提取物具有抗炎、抗癌、抗心力衰竭的作用,而其中的甾体类天然产物是其主要活性成分。bufogargarizin a具有罕见的[7-5-6-5]四环核心骨架,以及10个手性中心,具有非常高的氧化态,因此也导致其合成难度与合成价值都非常高。受到天然来源的限制,目前天然产物bufogargarizin a由自然界分离所获得的量很少,从而很难对其进行更深入的生物活性研究。
4、目前bufogargarizin a尚没有全合成方法的报道,基于此,开发一种bufogargarizin a合成方法对bufogargarizin a展开进一步活性研究具有重要的现实意义。
技术实现思路
1、基于此,本发明提供了一种bufogargarizin a的制备方法,其过程简洁高效,同时还可以为bufogargarizin a的结构修饰奠定基础。
2、本发明通过如下技术方案实现。
3、一种bufogargarizin a的制备方法,包括如下步骤:
4、将化合物1与二异丁基氢化铝在第一有机溶剂中进行还原反应,将还原反应的产物、叔丁基二甲基氯硅烷与碱在第二有机溶剂中进行羟基保护反应,制备化合物2将所述化合物2与氢化铝锂在第三有机溶剂中进行开环反应,将开环反应的产物、草酰氯、二甲基亚砜和三乙胺在第四有机溶剂中进行氧化反应,制备化合物3
5、将所述化合物3与环丙基溴化镁在第五有机溶剂中进行亲核加成反应,将亲核加成反应的产物、三乙基硅基三氟甲磺酸酯与n,n-二异丙基乙胺混合,进行硅基化反应,制备化合物4
6、将所述化合物4、对甲苯磺酰甲基异腈、叔丁醇钾与第六有机溶剂混合,进行范勒森反应,制备化合物5将所述化合物5与二异丁基氢化铝在第七有机溶剂中进行还原反应,然后将还原反应的产物与甲醇钠在第八有机溶剂中进行异构化反应,制备化合物6将所述化合物6、
7、(1-重氮基-2-氧代丙基)膦酸二甲酯与碳酸钾在所述第八有机溶剂中进行seyferth–gilbert增碳反应,然后将seyferth–gilbert增碳反应的产物与四丁基氟化铵进行脱硅反应,制备化合物7
8、将所述化合物7、戴斯-马丁氧化剂、碳酸氢钠与第九有机溶剂混合,进行氧化反应,制备化合物8
9、将所述化合物8、三甲基硅基三氟甲磺酸酯与二异丙基乙胺在第十有机溶剂中进行烯醇硅醚化反应,制备化合物9在三(乙腈基)环戊二烯六氟磷酸钌的催化作用下,将所述化合物9在第十一有机溶剂中进行[5+2]环加成反应,制备化合物10
10、在氧气氛围下,将所述化合物10、三苯基膦、乙酰丙酮锰、苯硅烷与第十二有机溶剂混合,进行水合反应,制备化合物11
11、将所述化合物11与二异丁基氢化铝在第十三有机溶剂中进行还原反应,将还原反应的产物与四丁基氟化铵进行脱硅反应,然后将脱硅反应的产物、戴斯-马丁氧化剂、碳酸氢钠与第十四有机溶剂混合,进行氧化反应,制备化合物12
12、将所述化合物12、双三甲基硅基胺基锂、三乙胺与第十五有机溶剂混合,进行烯醇化反应,然后将烯醇化反应的产物与三甲基氯硅烷进行硅基化反应,再将硅基化反应的产物与醋酸钯进行氧化反应,制备化合物13
13、将所述化合物13、二异丙基乙胺、硅胶与第十六有机溶剂混合,进行双键移位反应,制备化合物14
14、将所述化合物14、氢氧化钠水溶液与双氧水在第十七有机溶剂中进行环氧化反应,制备化合物15在氧气氛围下,将所述化合物15、三苯基膦、乙酰丙酮锰、苯硅烷与第十八有机溶剂混合,进行水合反应,将加成反应的产物、三甲基硅基咪唑、四丁基氟化铵与第十九有机溶剂混合,进行硅基化反应,制备化合物16
15、将所述化合物16、双三甲基硅基胺基钾、2-[n,正双(三氟甲烷烷磺酰)氨基]-5-氯吡啶与第二十有机溶剂混合,进行三氟甲磺酸烯酯化反应,制备化合物17
16、将所述化合物17与碘化钐在第二十一有机溶剂中进行开环反应,制备化合物18
17、将所述化合物18、氯甲基甲醚、4-二甲氨基吡啶、二异丙基乙胺与第二十二有机溶剂混合,进行羟基保护反应,将羟基保护反应的产物与二异丁基氢化铝在第二十三有机溶剂中进行还原反应,将还原反应的产物、三甲基氯硅烷与咪唑混合,进行硅基化反应,制备化合物19
18、将所述化合物19、与催化剂混合,在第二十四有机溶剂中进行铃木偶联反应,制备化合物20
19、将所述化合物20与间氯过氧苯甲酸在第二十五有机溶剂中进行环氧化反应,将环氧化反应的产物与硼烷二甲硫醚进行还原、双键迁移与开环氧串联反应,制备化合物21
20、将所述化合物21、戴斯-马丁氧化剂、碳酸氢钠与第二十六有机溶剂混合,进行氧化反应,制备化合物22
21、将所述化合物22与1,8-二偶氮杂双螺环[5.4.0]十一-7-烯在第二十七有机溶剂中进行消除反应,将消除反应的产物与双三甲基硅基氨基锂进行异构化反应,制备化合物23
22、将所述化合物23、(r)-2-甲基-cbs-恶唑硼烷、硼烷二甲硫醚与第二十八有机溶剂混合,进行还原反应,将还原反应的产物、4-二甲氨基吡啶、三乙胺与乙酸酐混合,进行乙酰化反应,制备化合物24
23、在氩气氛围下,将所述化合物24、氢氧化钠与n-溴代琥珀酰亚胺在第二十九有机溶剂中进行环氧化反应,同时部分脱硅,制备化合物25将所述化合物25与四丁基氟化铵进行脱硅反应,制备化合物26将所述化合物26、戴斯-马丁氧化剂、碳酸氢钠与第三十有机溶剂混合,进行氧化反应,制备化合物27
24、将所述化合物27与四氟硼酸锂在第三十一有机溶剂中进行脱保护反应,制备bufogargarizin a,所述bufogargarizin a的结构式如下所示:
25、
26、在其中一个实施例中,从所述化合物5制备所述化合物6的过程中满足如下条件中的一个或多个:
27、(1)所述化合物5与所述二异丁基氢化铝的摩尔比为1:(2.3~2.7);
28、(2)还原反应的温度为-78℃±5℃。
29、在其中一个实施例中,从所述化合物9制备所述化合物10的过程中满足如下条件中的一个或多个:
30、(1)[5+2]环加成反应的温度为55℃±5℃;
31、(2)[5+2]环加成反应的时间为12h±2h。
32、在其中一个实施例中,从所述化合物13制备所述化合物14的过程中满足如下条件中的一个或多个:
33、(1)所述化合物13、所述二异丙基乙胺与所述硅胶的摩尔比为1:(50~60):(14~15);
34、(2)双键移位反应的温度为25℃±5℃。
35、在其中一个实施例中,从所述化合物17制备所述化合物18的过程中满足如下条件中的一个或多个:
36、(1)所述化合物17与所述碘化钐的摩尔比为1:(2.3~2.7);
37、(2)开环反应的温度为-78℃±5℃。
38、在其中一个实施例中,从所述化合物19制备所述化合物20的过程中满足如下条件中的一个或多个:
39、(1)所述化合物19与的摩尔比为1:(1~1.4);
40、(2)铃木偶联反应的温度为60℃±5℃。
41、在其中一个实施例中,从所述化合物20制备所述化合物21的过程中满足如下条件中的一个或多个:
42、(1)所述化合物20、所述间氯过氧苯甲酸与所述硼烷二甲硫醚的摩尔比为1:(1~1.4):(1.3~1.7);
43、(2)环氧化反应的温度为25℃±5℃;
44、(3)还原、双键迁移与开环氧串联反应的温度为0℃±5℃。
45、在其中一个实施例中,从所述化合物22制备所述化合物23的过程中满足如下条件中的一个或多个:
46、(1)所述化合物22、所述1,8-二偶氮杂双螺环[5.4.0]十一-7-烯与所述双三甲基硅基氨基锂的摩尔比为1:(1.8~2.2):(3.8~4.2);
47、(2)消除反应的温度为25℃±5℃;
48、(3)异构化反应的温度为-98℃±5℃。
49、在其中一个实施例中,从所述化合物24制备所述化合物25的过程中满足如下条件中的一个或多个:
50、(1)所述化合物24、所述氢氧化钠与所述n-溴代琥珀酰亚胺的摩尔比为1:(1.8~2.2):(1~1.4);
51、(2)环氧化反应的温度为25℃±5℃。
52、在其中一个实施例中,从所述化合物27制备所述bufogargarizin a的过程中满足如下条件中的一个或多个:
53、(1)所述化合物27与所述四氟硼酸锂的摩尔比为1:(9~11);
54、(2)脱保护反应的温度为82℃±5℃。
55、与现有技术相比较,本发明的bufogargarizin a的制备方法具有如下有益效果:
56、本发明以为起始物,提供了一种天然产物bufogargarizin a的全合成方法,制备过程简洁高效,收率高,副产物少,能够大幅度提高bufogargarizin a的获得量,从而能够对bufogargarizin a进行更深入的生物活性研究。此外,本发明的bufogargarizin a的制备方法较易对其结构进行修饰,从而能够从中筛选出一系列先导化合物来研究其生物活性,进而对抗炎、抗肿瘤等人类重大疾病的研究方向作出贡献。
- 还没有人留言评论。精彩留言会获得点赞!