一种碳键催化剂及其制备方法与应用
本发明涉及一种碳键催化剂及其制备方法与应用,属于有机合成。
背景技术:
1、碳元素在有机化学中占据着中心地位,在以碳为中心的化学反应过程中,理论上会形成定向的碳键弱相互作用,用碳键作用来催化化学反应不仅能够为化学物质的合成提供潜在的新催化策略,而且对于理解反应中间体的活性以及反应的各种选择性具有重要意义。山东大学王瑶课题组致力于非传统弱相互作用(σ-hole以及π-hole)催化反应的研究,以前期研究为基础,该课题组提出了碳键催化模式,即利用催化剂碳中心和电子供体形成的定向弱相互作用来驱动化学反应(angew.chem.int.ed.2021,60,22717–22721)。作者验证了碳键催化的可行性,并通过不同类型的反应证明了这一催化模式的一般性和潜力。
2、超分子碳键催化作为一种刚被证实的弱相互作用催化模式,前期研究证实这类碳键催化剂具有双活化位点并相比氢键类催化剂拥有更强的方向性,这些独特的化学性质有望使碳键可以实现一些传统方法难以解决的催化难题,所以开展对碳键催化的研究是至关重要的。对于已发展的碳键催化剂来说,其溶解度较差、催化效率较低使其在具体应用中具有一定的局限性。因此,构建其他骨架的碳键催化剂十分关键。巴比妥酸具有较强的酸性,并且在n原子上可以引入修饰集团,将其代替米氏酸引入碳键催化剂中,可以很好的改善溶解度差、效率低等问题,大大提高碳键催化剂在有机合成当中的应用范围,具有很大的应用潜力。为此,提出本发明。
技术实现思路
1、针对现有技术的不足,本发明提供了一种碳键催化剂及其制备方法与应用。本发明以手性伯胺为原料,与异氰酸酯或固体光气反应,先合成一系列巴比妥酸的衍生物,再继续与四氰基乙烯反应,合成新型碳键催化剂。本发明的反应条件温和,对环境无污染。此外,该合成方法的成本低,反应的产率和优异,为手性碳键催化剂的开发利用提供了技术支持。
2、术语解释:
3、室温:具有本领域公知含义,指25±5℃。
4、本发明的技术方案如下:
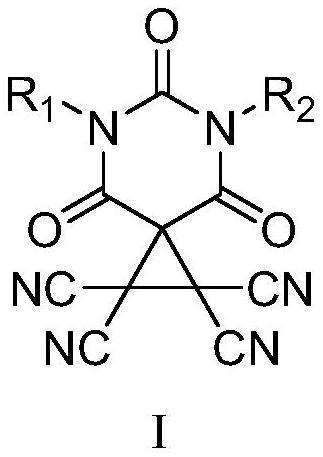
5、一种碳键催化剂,所述碳键催化剂具有如下式i所示结构:
6、
7、式i中,r1为苯乙基、苄基、
8、r2为
9、根据本发明优选的,式i中,当r1为苯乙基或苄基时,r2为当r1为时,r2与r1相同。
10、本发明中*代表手性碳原子。
11、根据本发明,上述碳键催化剂的制备方法,包括步骤如下:
12、(1)以手性伯胺v为起始原料,与异氰酸酯类化合物vi或固体光气反应,得到中间体ii;
13、
14、式v中,r3为
15、式vi中,r4为苯乙基、苄基、
16、式ii中,r1、r2与式i中相同;
17、(2)在氩气保护下,中间体ii与丙二酰氯进行缩合关环反应,得到中间体iii;
18、
19、式iii中,r1、r2与式ii中相同;
20、(3)在氩气保护下,在碱存在下,中间体iii与br2经取代反应,得到中间体iv;
21、
22、式iv中,r1、r2与式iii中相同;
23、(4)在氩气保护下,中间体iv与四氰基乙烯经环化反应,得到碳键催化剂i。
24、根据本发明优选的,步骤(1)中,手性伯胺v与异氰酸酯类化合物vi反应的具体步骤为:将异氰酸酯类化合物vi加入二氯甲烷中,之后加入手性伯胺v,室温下反应1-3小时;反应完成后,经后处理,得到中间体ii;
25、进一步优选的,所述异氰酸酯类化合物vi与手性伯胺v的摩尔比为1-1.2:1;所述二氯甲烷的体积与异氰酸酯类化合物vi的摩尔数之比为0.5-5ml:1mmol。
26、进一步优选的,所述后处理的步骤为:向所得反应液中加入二氯甲烷2倍体积的1mol/lhcl溶液,之后使用二氯甲烷萃取,将所得有机相依次使用饱和氯化钠溶液、水洗涤后,将所得有机相用无水硫酸钠干燥,之后过滤,除去溶剂,得到中间体ii。
27、根据本发明优选的,步骤(1)中,手性伯胺v与固体光气反应的具体步骤为:氩气保护下,向固体光气的二氯甲烷溶液中加入手性伯胺v和n,n’-二异丙基乙胺进行第一反应,生成异氰酸酯化合物;向第一反应所得反应液中,继续加入手性伯胺v和n,n’-二异丙基乙胺,进行第二反应;反应完成后,经后处理步骤,得到中间体ii;
28、进一步优选的,所述固体光气的二氯甲烷溶液的浓度为0.1-0.3mmol/ml;
29、进一步优选的,进行第一反应时,原料比例如下:所述固体光气与手性伯胺v的摩尔比为0.33-0.4:1;所述n,n’-二异丙基乙胺与手性伯胺v的摩尔比为1-2.2:1;进行第一反应时,所述手性伯胺v是以手性伯胺v的二氯甲烷溶液的形式加入的,所述手性伯胺v的二氯甲烷溶液的浓度为0.1-0.5mmol/ml;
30、进一步优选的,所述第一反应的时间为5-10min;
31、进一步优选的,进行第二反应时,所加入的手性伯胺v和n,n’-二异丙基乙胺的量与第一反应时相同;所述手性伯胺v和n,n’-二异丙基乙胺是以手性伯胺v和n,n’-二异丙基乙胺混合物的二氯甲烷溶液的形式加入体系中,混合物的二氯甲烷溶液中手性伯胺v的浓度为0.1-0.5mmol/ml;
32、进一步优选的,所述第二反应的时间为10-15min;
33、进一步优选的,所述后处理步骤为:用质量分数为10%的khso4水溶液猝灭反应;之后用乙酸乙酯萃取,所得有机相依次用质量分数为5% nahco3水溶液和饱和氯化钠溶液洗涤,之后所得有机相用na2so4干燥,抽滤、除去溶剂,所得粗产物经柱层析分离纯化,得中间体ii,柱层析所用洗脱剂为石油醚和乙酸乙酯的混合溶剂,混合溶剂中石油醚和乙酸乙酯的体积为4-6:1。
34、根据本发明优选的,步骤(2)中所述中间体ii与丙二酰氯的摩尔比为1:1-1.2;所述丙二酰氯滴加入体系中,滴加时间为5-10min。
35、根据本发明优选的,步骤(2)中所述缩合关环反应于二氯甲烷中进行,所述二氯甲烷的体积与中间体ii的摩尔数之比为30-50ml:1mmol。
36、根据本发明优选的,步骤(2)中所述缩合关环反应的时间为4-6小时。
37、根据本发明优选的,步骤(2)中,缩合关环反应完成后,还包括后处理步骤,具体如下:将缩合关环反应完成后所得反应液依次用饱和氯化钠溶液、水洗涤,所得有机相用无水硫酸钠干燥,抽滤、除去溶剂,所得粗产物经柱层析分离纯化后,得中间体iii,柱层析所用洗脱剂为石油醚和乙酸乙酯的混合溶剂,混合溶剂中石油醚和乙酸乙酯的体积为4-6:1。
38、根据本发明优选的,步骤(3)中所述碱为naoh,所述碱与中间体iii的摩尔比为1-1.2:1。
39、根据本发明优选的,步骤(3)中所述中间体iii与br2的摩尔比为1:1-1.2。
40、根据本发明优选的,步骤(3)中所述取代反应于水和乙腈的混合溶剂中进行,混合溶剂中水与乙腈的体积比为1:1;所述混合溶剂的体积与中间体iii的摩尔数之比为3-5ml:1mmol。
41、根据本发明优选的,步骤(3)中所述取代反应的温度为0-5℃,所述取代反应的时间为10-15min。
42、根据本发明优选的,步骤(3)中所述取代反应的具体步骤为:在氩气保护下,将中间体iii加入水和乙腈的混合溶剂中,之后加入碱,在0-5℃下,向体系中滴加br2,滴加速率为1滴/s,滴加完成后,0-5℃下反应10-15min。
43、根据本发明优选的,步骤(3)中,取代反应完成后,还包括后处理步骤,具体如下:将取代反应所得反应液用二氯甲烷萃取,将所得有机相用无水硫酸钠干燥,抽滤、除去溶剂,得中间体iv。
44、根据本发明优选的,步骤(4)中所述中间体iv与四氰基乙烯的摩尔比为1:1-1.2;所述中间体iv的摩尔数以中间体iii计。
45、根据本发明优选的,步骤(4)中所述环化反应的时间为5-8分钟。
46、根据本发明优选的,步骤(4)中所述环化反应于1,4-二氧六环中进行,所述1,4-二氧六环的体积与中间体iv的摩尔数之比为1-3ml:1mmol。
47、根据本发明优选的,步骤(4)中,环化反应完成后,还包括后处理步骤,具体如下:在搅拌下将碎冰加入所得反应液中,搅拌直到冰完全融化,向体系中加入二氯甲烷萃取,所得有机相用水洗涤后用无水硫酸钠干燥,抽滤、除去溶剂后,加入正己烷搅拌,有固体析出,抽滤,所得固体用正己烷洗涤,干燥,得碳键催化剂i;所述碎冰的质量与中间体iv的摩尔数之比为2-5g:1mmol;所述正己烷与1,4-二氧六环的体积比为1-4:1。
48、根据本发明优选的,上述碳键催化剂在催化氧杂pictet-spengler反应中的应用。
49、本发明的技术特定及有益效果如下:
50、1、本发明以手性伯胺为原料,将其与异氰酸酯或固体光气反应,先合成一系列巴比妥酸的衍生物,再继续与四氰基乙烯反应,合成新型碳键催化剂。本发明的反应条件温和,操作简单,便于碳键催化剂的大量制备。此外,可以从廉价的手性原料出发高效构建一类手性碳键骨架,本发明大大拓展了之前碳键催化剂骨架难以修饰的难题,为后期碳键催化剂的合理设计提供了新思路。具体研究表明本发明合成的新型碳键催化剂相比传统碳键催化剂溶解度更好,催化效率更高,为碳键催化的具体应用提供有力保障。
51、2、本发明合成的碳键催化剂溶解度高,催化效率大大提高,大大提高碳键催化剂在有机合成当中的应用范围。
- 还没有人留言评论。精彩留言会获得点赞!