一种索托拉西布关键中间体的制备方法与流程
本发明属于化学合成,具体地说是一种抗癌药物索托拉西布关键中间体2-异丙基-3-氨基-4-甲基吡啶的制备方法。
背景技术:
1、2-异丙基-3-氨基-4-甲基吡啶是制备lumakras(sotorasib)的关键中间体。lumakras(sotorasib)索托拉西布,前称amg 510,是一种小分子,旨在与kras g12c结合,将蛋白质锁定在非活性状态,防止其发送驱动不受控制的细胞生长的信号。该药物的靶向方法不会影响未突变的kras蛋白。2021年05月28日,安进(amgen)宣布美国fda已加速批准kras g12c抑制剂lumakras(sotorasib)上市,用于治疗既往至少接受过一次系统治疗的携带kras g12c突变局部晚期或转移性非小细胞肺癌(nsclc)患者。
2、文献报道的2-异丙基-3-氨基-4-甲基吡啶的制备方法主要有:
3、1)专利wo2021097207提出以2-氯-3-氨基-4甲基吡啶为起始原料,经铃木偶联反应,再经过一步还原得目标产物,反应式如下:
4、
5、该方法起始原料价格高,并且用了贵金属催化,所用到的硼酸试剂也比较昂贵,所以该方法不是此中间体合成的最佳方案。
6、2)专利wo2021097207同时也报道了异丁酸乙酯为起始原料,经成环,水解,hofmann重排反应最后得到目标产物,反应式如下:
7、
8、该方法路线原料比较便宜易得,但是该方案由于氰基为易水解基团,较为不稳定,所以限制了该项目的产业化过程,而且该专利也没有提及具体的后处理方式和满意的收率,所以对其产业化可行性还是有一定的未知度。
技术实现思路
1、为了克服现有技术的缺点和不足,本发明提供了一种操作简单、原料便宜,中间体稳定、反应污染小、产物收率高的索托拉西布关键中间体2-异丙基-3-氨基-4-甲基吡啶的制备方法。
2、本发明所采取的技术方案为:
3、一种索托拉西布关键中间体的制备方法,以乙酰乙酸乙酯为原料,先和异丁酰氯生成异丁酰乙酸乙酯,再经过取代,成环,氨解,hofmann重排制得目标产物2-异丙基-3-氨基-4-甲基吡啶。
4、具体反应方程式如下:
5、
6、作为优选,所述索托拉西布关键中间体的制备方法,包括下述步骤:
7、(1)在反应瓶中加入乙酰乙酸乙酯,再加入甲苯、乙醇镁,升温反应1小时,最后滴加异丁酰氯,再反应2~6小时,经后处理得到产物异丁酰乙酸乙酯;
8、(2)在反应瓶中加入异丁酰乙酸乙酯、丙酮、l-脯氨酸,升温反应,保温2~6小时,检测原料反应完全后,经后处理得到2-异丁酰基-3-甲基-2-丁烯腈;
9、(3)在反应瓶中加入2-异丁酰基-3-甲基-2-丁烯腈,加入乙醇、dmf-dma,升温反应2小时,检测原料反应完毕后,再加入乙酸铵,加入氨水,升温回流2~8小时,检测中间体消失后,蒸干乙醇,加入水和二氯甲烷,二氯甲烷用饱和氯化钠洗涤,干燥后,旋干,最后得到产品2-异丙基-4-甲基烟酸乙酯;
10、(4)在反应瓶中加入2-异丙基-4-甲基烟酸乙酯、乙醇、氨水,升温反应过夜,经后处理后得到2-异丙基-4-甲基烟酰胺;
11、(5)在反应瓶中加入水、氢氧化钾,加入2-异丙基-4-甲基烟酰胺,搅拌下滴加溴素,保持滴加温度在t=0-10℃之间,检测中控原料消失后,升温反应2~4小时,检测原料和中间态消失,经后处理后得到2-异丙基-3-氨基-4-甲基吡啶。
12、作为优选,步骤(1)中,乙酰乙酸乙酯、异丁酰氯、乙醇镁物质的量之比为1:1~1.5:1~2,反应温度为60~80℃;步骤(1)中后处理操作为:滴加盐酸淬灭反应,有机相加入氨水,1~10℃搅拌1小时,分出有机相,用醋酸调节ph=7,搅拌1小时,分出有机相,蒸出甲苯,精馏。
13、作为优选,步骤(2)中,异丁酰乙酸乙酯、l-脯氨酸物质的量之比为1:0.1~2,反应温度为30℃~回流;步骤(2)中后处理操作为:降温,加入盐酸调节ph=3~4,过滤,有机相旋蒸得到产品,直接进行下一步骤。
14、作为优选,步骤(3)中,2-异丁酰基-3-甲基-2-丁烯腈、dmf-dma、乙酸铵、氨水物质的量之比为1:1~3:0.1~2:1~3,反应温度为30℃~回流。
15、作为优选,步骤(4)中,2-异丙基-4-甲基烟酸乙酯、氨水物质的量之比为1:1~15,反应温度为20~50℃;步骤(4)中后处理操作为:析出固体,抽滤。
16、作为优选,步骤(5)中,2-异丙基-4-甲基烟酰胺、氢氧化钾、溴素,物质的量之比为1:1~5:1~3,滴加温度在0~10℃之间,反应温度为40~80℃。
17、作为优选,步骤(5)中后处理操作为:降温至室温,加ea搅拌5min,分层,水相再用ea萃取一次,合并有机相,加入活性炭,升温至回流脱色,过滤,减压蒸馏,得到粗品,粗品用环己烷降温打浆,过滤得到2-异丙基-3-氨基-4-甲基吡啶。
18、作为优选,所述索托拉西布关键中间体的制备方法,具体包括下述步骤:
19、(1)在500ml反应瓶中加入乙酰乙酸乙酯0.77mol,再加入甲苯600g、乙醇镁0.92mol,升温至60℃反应1小时,最后滴加异丁酰氯0.85mol,再升温至t=90℃反应4小时,滴加100g盐酸淬灭反应,有机相用加入氨水150g,1~10℃搅拌1小时,分出有机相,用醋酸调节ph=7,搅拌1小时,分出有机相,蒸出甲苯,精馏后得到产物异丁酰乙酸乙酯;
20、(2)在反应瓶中加入异丁酰乙酸乙酯0.63mol、丙酮600g、l-脯氨酸0.13mol,升温至回流反应,保温5小时,检测原料反应完全后,降温,加入盐酸调节ph=3-4,过滤,有机相旋蒸得到2-异丁酰基-3-甲基-2-丁烯腈;
21、(3)在反应瓶中加入上步2-异丁酰基-3-甲基-2-丁烯腈0.45mol,加入乙醇450g,dmf-dma 0.68mol,升温回流反应2小时,检测原料反应完毕后,再加入乙酸铵0.09mol,加入氨水1.13mol,升温回流8小时,检测中间体消失后,蒸干乙醇,加入300水和300g二氯甲烷,二氯甲烷用饱和氯化钠洗涤,干燥后,旋干,最后得到产品2-异丙基-4-甲基烟酸乙酯;
22、(4)在反应瓶中加入2-异丙基-4-甲基烟酸乙酯0.41mol,乙醇300g,氨水3.3mol,升温至40℃反应过夜,降至室温析出固体,抽滤,得到2-异丙基-4-甲基烟酰胺;
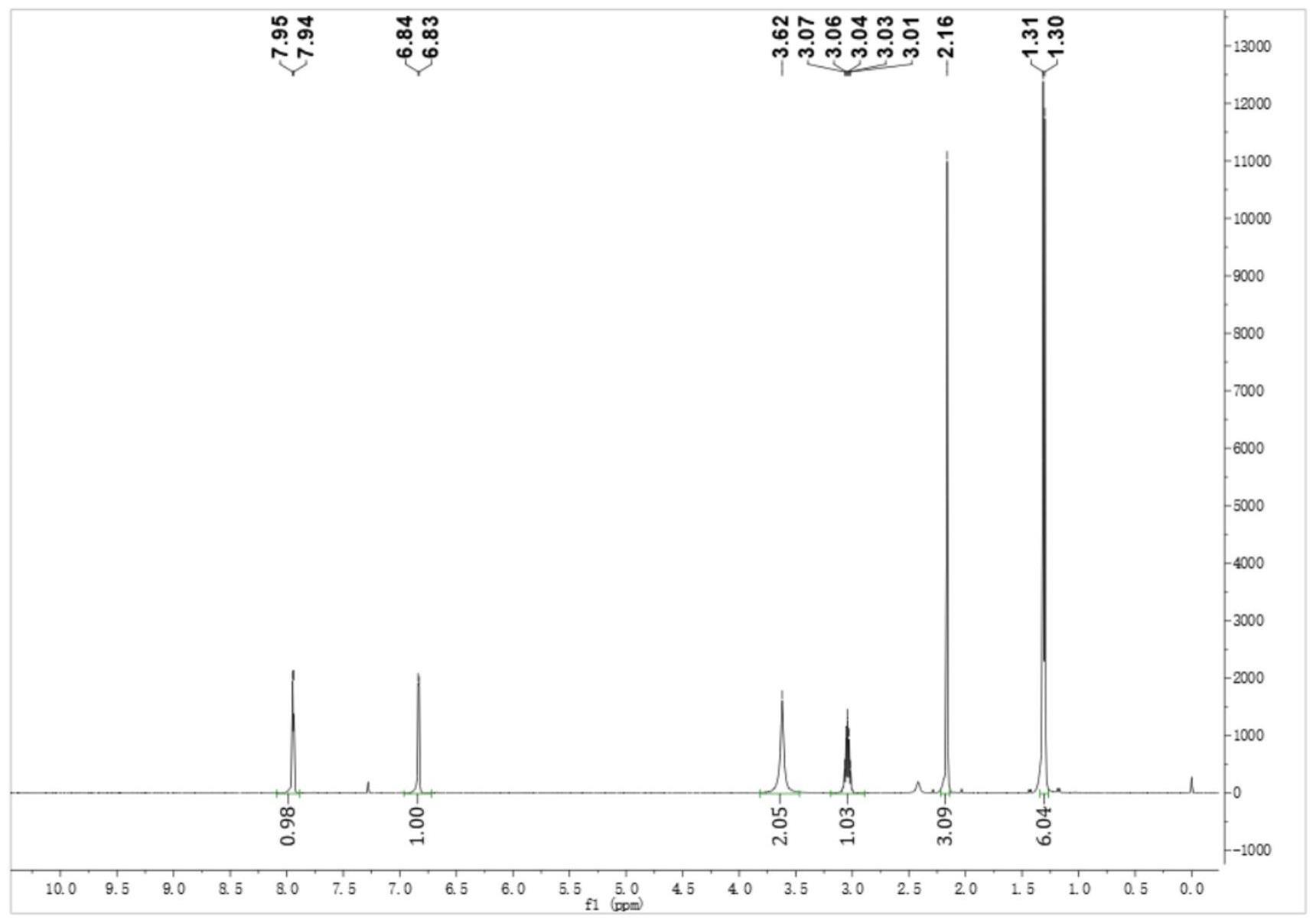
23、(5)在反应瓶中加入水600g,氢氧化钾0.79mol,加入2-异丙基-4-甲基烟酰胺0.31mol,搅拌下滴加溴素0.38mol,保持滴加温度在t=0~10℃之间,检测中控原料消失后,快速升温60℃保温反应2小时,检测原料和中间态消失,降温至室温,加300g乙酸乙酯搅拌5min,分层,水相再用100g乙酸乙酯萃取一次,合并有机相,加入2g活性炭,升温至回流脱色,过滤,减压蒸馏,得到粗品,粗品用环己烷降温打浆,过滤得到精品2-异丙基-3-氨基-4-甲基吡啶。
24、与现有技术相比,本发明具有以下优势:
25、(1)本发明2-异丙基-3-氨基-4-甲基吡啶的制备过程中原料便宜易得,没有高污染和高危险性的溶剂或催化剂,后处理较简单,产品纯度高,收率高,操作简单,污染少,成本低;
26、(2)本发明所有的反应均为常见的反应,稳定安全,所有的中间体基团稳定性好,是更适合该产品的产业化的路线。
- 还没有人留言评论。精彩留言会获得点赞!