一种尼拉帕利杂质1的制备方法及其应用与流程
1.本发明涉及化学合成技术领域,尤其涉及一种尼拉帕利杂质1的制备方法及其应用。
背景技术:
2.尼拉帕利是一种口服的聚adp核糖聚合酶(parp)抑制剂,抑制细胞对dna损伤的修复,对于带有brca基因突变的癌细胞来说,倘若parp活性进一步受到抑制,这些细胞分裂时就会产生大量的dna损伤,导致癌细胞死亡。尼拉帕利,开发适应症为卵巢癌、乳腺癌、前列腺癌等,该化合物对上病症,具有良好治疗效果。其化学结构式为:
3.本发明目标化合物,是尼拉帕利的一种重要杂质,欧洲药典收录为杂质1(以下称为“尼拉帕利杂质1”)。该化合物的合成制备目前无相关文献报道,不能为尼拉帕利的质量控制相关研究提供廉价易得、高质量的杂质对照品。
技术实现要素:
4.本发明的目的是提供一种尼拉帕利杂质1的制备方法及其应用,解决了现有技术中由于制备技术空白而不能为尼拉帕利的质量控制提供基础的问题。
5.为了实现上述目的,本发明采用了如下技术方案:第一方面,本发明提供了一种尼拉帕利杂质1的制备方法,包括如下步骤:将式i所示结构化合物溶于有机溶剂中,加入酸和水后与水合氯醛、盐酸羟胺在加热条件下进行反应,得到式ii所示结构化合物,即尼拉帕利杂质1;其中,式i所示结构化合物、式ii所示结构化合物如下所示:
6.进一步,尼拉帕利杂质1的制备方法,包括如下步骤:将式i所示结构化合物溶于有机溶剂中,加入酸和水后与水合氯醛、盐酸羟胺在加
热条件下进行反应,反应结束后,旋蒸干有机溶剂,柱层析纯化,得到式ii所示结构化合物。
7.作为具体的技术方案,所述有机溶剂优选为水、乙腈、2-丁酮、乙腈、2-甲基四氢呋喃中的任意一种。
8.作为具体的技术方案,式i所示结构化合物的反应浓度为0.1~0.35摩尔/升。
9.作为具体的技术方案,所述酸为对甲苯磺酸、硫酸中的任意一种,其使用量为式i所示结构化合物的摩尔量的0.5~1.5当量。
10.作为具体的技术方案,水合氯醛参与反应的量为式i所示结构化合物的摩尔量的1~1.5当量。
11.作为具体的技术方案,盐酸羟胺参与反应的量为式i所示结构化合物的摩尔量的2~4当量。
12.作为具体的技术方案,反应温度为70~100℃。
13.作为具体的技术方案,柱层析使用流动相为石油醚与乙酸乙酯混合流动相,其中石油醚与乙酸乙酯的体积比为1:0.15~0.4。
14.第二方面,本发明提供了由上述的制备方法得到的尼拉帕利杂质1在制备尼拉帕利杂质对照品中的应用。
15.与现有技术相比,本发明提供了一种尼拉帕利杂质1的制备方法及其应用,具备以下有益效果:本发明提供了一种新的合成路线,以2-氨基-3-甲基苯甲酸甲酯为原料,一步即可得到尼拉帕利杂质1,本发明制备方法原料易得,步骤少,产率高,成本低廉,填补了尼拉帕利杂质1制备的技术空白,可为尼拉帕利质量研究提供廉价、高质量的杂质对照品,对尼拉帕利安全用药有重要意义。
附图说明
16.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
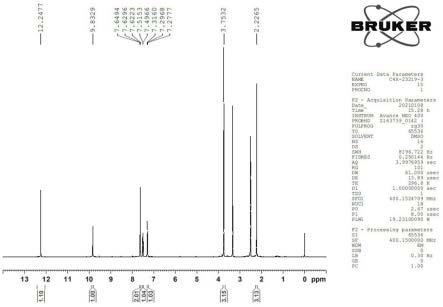
17.图1为本发明实施例1合成的产物一维核磁氢谱;图2为本发明实施例1合成的产物质谱图;图3为本发明实施例1合成的产物高效液相图谱。
具体实施方式
18.下面将对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
19.本发明提供了一种尼拉帕利杂质1的制备方法,本发明是以式i所示结构化合物为起始原料合成式ii所示结构化合物。如未明确指出,本发明所使用的试剂均为购自市场的常用试剂,所涉及操作温度,如无说明,均为室温25℃条件下进行。
20.其中,式i所示结构化合物、式ii所示结构化合物如下:
21.式i所示结构化合物为2-氨基-3-甲基苯甲酸甲酯,为商业途径可获得原料;其他原料包括水合氯醛以及盐酸羟胺均可通过商业途径获得;式ii所示结构化合物,为尼拉帕利杂质1。
22.本发明的合成反应过程如下所示:
23.下面通过详细的实施例并结合附图对本发明作进一步详细描述。
实施例1
24.本实施例提供了尼拉帕利杂质1的合成制备情况,具体为:于250ml干燥的三口瓶中,量取80ml水和1.6g浓硫酸,再依次加入2g的2-氨基-3-甲基苯甲酸甲酯、2.2g的水合氯醛和1.85g的盐酸羟胺。混合物加热至95℃搅拌1h。监测反应完成后,终止反应,旋蒸干有机溶剂,柱层析纯化,流动相为石油醚:乙酸乙酯=1:0.3,得到米色粉末状固体2.4g(收率84%),即为尼拉帕利杂质1。尼拉帕利杂质1谱图数据如下:1h-nmr (dmso-d6, 400mhz.): δ 12.25 (s, 1h), 9.83 (s, 1h), 7.64 (dd, j = 7.8, 1.5 hz, 1h), 7.62 (s, 1h), 7.54
ꢀ–ꢀ
7.47 (m, 1h), 7.30 (t, j = 7.7 hz, 1h), 3.75 (s, 3h), 2.23 (s, 3h). lcms:(m/z)237[m+1]
+
。
[0025]
图1展示了合成的产物一维核磁氢谱。图2展示了合成的产物质谱图,图中各峰的数值从左至右依次为:87、146、147、237、278、300、341、424、465、513、535、625、666、712、730、783、826、889、937、968。图3展示了合成的产物高效液相图谱。
实施例2
[0026]
本实施例提供了尼拉帕利杂质1的合成制备情况,具体为:于250ml干燥的三口瓶中,量取80ml乙腈和1.6g浓硫酸,再依次加入2g的2-氨基-3-甲基苯甲酸甲酯、2.6g的水合氯醛和2.0g的盐酸羟胺。混合物加热至80℃搅拌1h。监测反应完成后,终止反应,旋蒸干有机溶剂,柱层析纯化,流动相为石油醚:乙酸乙酯=1:0.3,得到米色粉末状固体2.0g(收率70%),即为尼拉帕利杂质1。
实施例3
[0027]
本实施例提供了尼拉帕利杂质1的合成制备情况,具体为:
于250ml干燥的三口瓶中,量取80ml水和2.3g对甲苯磺酸,再依次加入2g的2-氨基-3-甲基苯甲酸甲酯、2.2g的水合氯醛和1.85g的盐酸羟胺。混合物加热至90℃搅拌1h。监测反应完成后,终止反应,旋蒸干有机溶剂,柱层析纯化,流动相为石油醚:乙酸乙酯=1:0.3,得到米色粉末状固体2.5g(收率87%),即为尼拉帕利杂质1。
[0028]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
[0029]
此外,应当理解,虽然本说明书按照实施方式加以描述,但并非每个实施方式仅包含一个独立的技术方案,说明书的这种叙述方式仅仅是为清楚起见,本领域技术人员应当将说明书作为一个整体,各实施例中的技术方案也可以经适当组合,形成本领域技术人员可以理解的其他实施方式。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1