消旋的喜树碱中间体三元环化合物的合成方法与流程
本发明属于有机化学合成领域,具体涉及一种消旋的喜树碱中间体三元环化合物的合成方法,即4-乙基-7,8-二氢-ih-吡喃并[3,4-f]氮茚-3,6,10(4h)-三酮的合成方法。
背景技术:
1、喜树碱(r1=r2=h,camptothecin,cpt)是1966年由美国wall等(wall m e,wanim c,cook c e et al.j am chem soc.1966,88,3888-3890)首次从中国中部的珙桐科植物喜树中分离得到的一个含有喹啉五环骨架结构的生物碱。由于它在体内和体外的动物实验中表现出显著的抗肿瘤活性,因此成为抗肿瘤药物研宄的重点之一。从1985年hsiang等(hsiang y h,hertzberg r,hecht s et al.j bio chem.1985,260,14873-14878)发现喜树碱是拓扑异构酶i的特异性抑制剂这一独特作用机制后,此类化合物的研发进入了高速发展阶段。
2、二十多年来喜树碱及其衍生物的高效化学全合成一直是药物化学研究领域的热点之一,国内外的不同研究小组已相继报道了30多条喜树碱及其类似物的全合成路线(duw.tetrahedron,2003,59:5120)。在众多合成策略中friedlander缩合法是用a环和cde环进行缩合来得到喜树碱,在喜树碱的结构改造中有着独到的优势,目前喜树碱的cde环可以由消旋的cde环经过手性拆分获得,而消旋的喜树碱及其衍生物也是喜树碱生物活性测试的重要对照品。
3、对于喜树碱的消旋cde环合成路线共有三条,但这些路线都存在着一些问题:
4、1)j.med.chem.1980,23,554-560提供了以下合成路线:
5、
6、该路线缺点如下:1.第一步和第四步的收率都比较低,且第一步反应时间太长;2.反应过程中使用的雷尼镍,处理较为繁复;3.乙基化反应需要低温操作,且反应时间较长;4.起始原料乙基5-氰基-4-甲基-6-氧亚基-1,6-二氢吡啶-2-甲酸基酯价格贵。
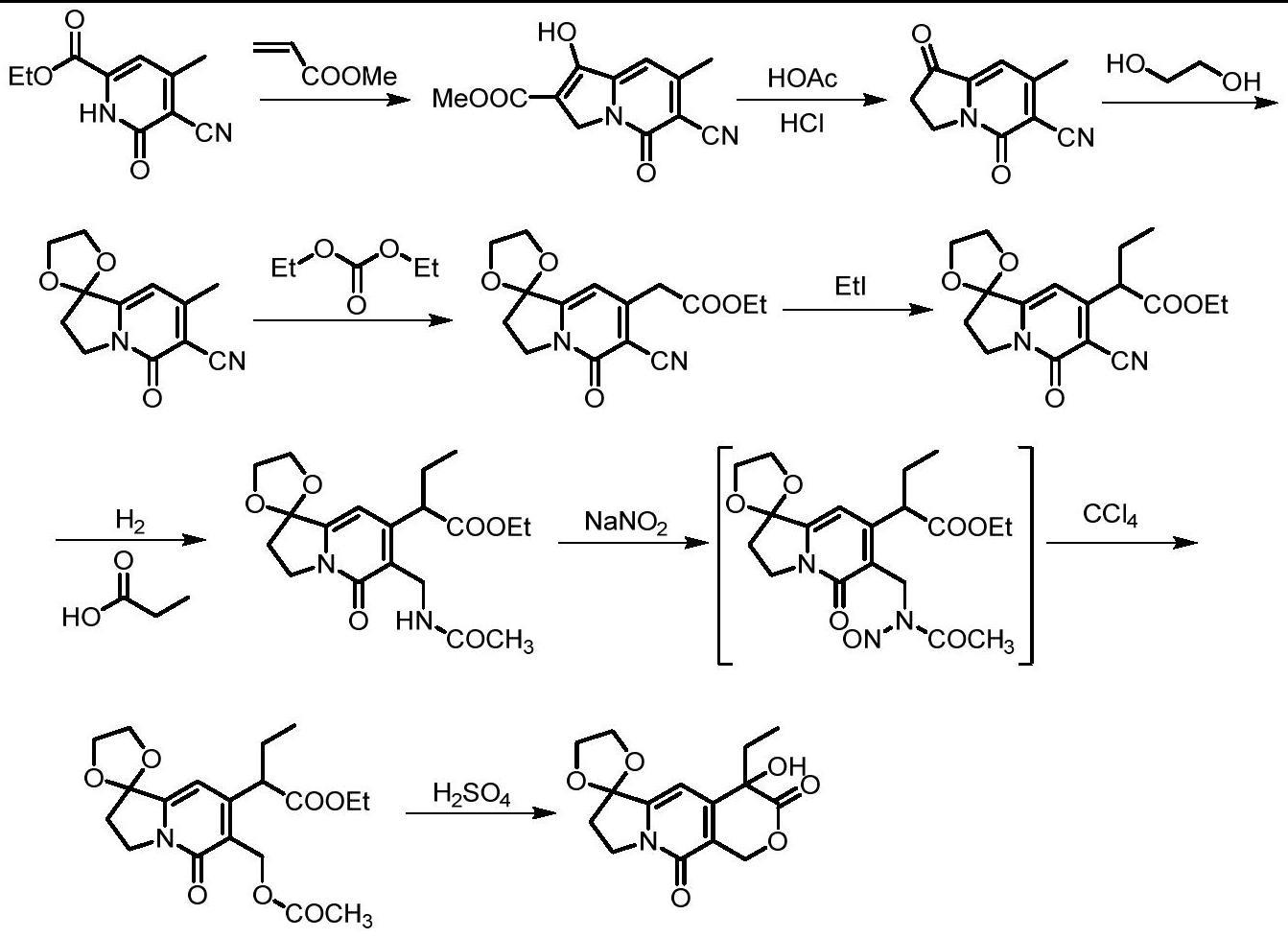
7、2)j.0rg.chem.l993,58,611-617提供了以下合成路线:
8、
9、该路线的缺点如下:1.该方法中氢溴酸脱羧的收率太低;2.氧化的条件苛刻并且收率低;3.反应过程中会产生毒性较高的废料,增加处理废料成本;4.起始原料烯基氯成本高。
10、3)current organic synthesis.2020,17,5880591提供了以下合成路线:
11、
12、该路线缺点如下:1.合成所用的起始原料价格高;2.反应过程中使用过渡金属催化剂都较为昂贵;3.反应过程中使用有毒气体一氧化碳,不利于大量生产。
13、综上所述,合成抗肿瘤药物喜树碱关键中间体三元环化合物即4-乙基-7,8-二氢-ih-吡喃并[3,4-f]氮茚-3,6,10(4h)-三酮目前的合成路线从成本和生产可行性来具有较大的优化空间和发展潜力。
技术实现思路
1、本发明针对现有的合成4-乙基-7,8-二氢-ih-吡喃并[3,4-f]氮茚-3,6,10(4h)-三酮路线的安全性低、收率低、原料成本高等问题,提供一种合成4-乙基-7,8-二氢-ih-吡喃并[3,4-f]氮茚-3,6,10(4h)-三酮的新方法。
2、具体技术方案如下:
3、1)向反应釜中加入氯代试剂(1.1-1.2mol)和二氯甲烷,后在氮气保护下降温到0-5℃,保温滴加下列反应线路中的式1化合物(1.0mol),滴加完毕,继续在0-5℃搅拌下缓慢滴加三乙胺(15-18mol),滴加完毕,在25-30℃下搅拌反应1.5h;后加入二氯甲烷萃取,有机相合并后浓缩;浓缩过滤,浓缩物用装有硅胶的砂芯漏斗过滤,用石油醚和乙酸乙酯的混合液(pe/ea=5/1)淋洗,滤液浓缩后得到式3化合物。
4、2)向反应釜中加入式2化合物(1.0mol),加入甲苯,加入硫酸二甲酯(1.0-1.1mol),80-90℃搅拌反应12h,再加入胺(1.1-1.2mol)和具有活性亚甲基的化合物(1.0mol);蒸馏除去甲苯,随后加入甲醇,加入甲醇钠,回流搅拌反应2h后,蒸馏除去甲醇,待反应冷却至室温,向瓶中加入冰水,用盐酸溶液调节ph至5-6;用二氯甲烷萃取有机相,无水硫酸钠干燥,浓缩后,用石油醚重结晶得到式4化合物;
5、3)在反应釜加入式3化合物(1.0mol)、式4化合物(0.75-0.80mol)、胺(5.0-6.0mol)、乙醇,在25-30℃反应12h;反应液过滤,滤饼用石油醚洗涤得到式5化合物;
6、4)在反应釜中加入式5化合物(1.0mol)和乙二醇二甲醚,降温至0-5℃加入叔丁醇钾(1.0-1.1mol),反应30min,加入乙基化试剂(1.8-2.0mol),25-30℃反应6h;反应用二氯甲烷萃取,有机相用无水硫酸钠干燥,浓缩得到式6化合物;
7、5)在加压釜加入式6化合物(1.0mol)、1,4-二氧六环、多聚甲醛(2.0-2.5mol)、酸(0.02-0.025体积比)、水(0.2-0.25体积比),在105-107℃反应24h;反应用二氯甲烷萃取,有机相用无水硫酸钠干燥,浓缩得到式7化合物;
8、6)在反应釜中加入式7化合物(1.0mol)、苯甲醛(1.0-1.1mol)、催化剂、dmf、thf的混合溶剂,-40℃~-50℃冷却并搅拌10分钟,加入双(三甲基硅基)氨基锂(1.1-1.2mol),在室温下反应24h,加入盐酸溶液继续搅拌2h,调节ph值为5-6,用二氯甲烷与甲醇的混合溶剂萃取。有机相用无水硫酸钠干燥、过滤、浓缩;浓干物加至酸溶液中,在90±2℃反应3h;反应加入碳酸钠中和,反应液中二氯甲烷萃取,有机相用无水硫酸钠干燥后浓缩,得到式8化合物;
9、7)在反应釜中加入式8化合物(1.0mol)、二氯甲烷、甲醇溶液,降温至-70℃~-75℃通入过氧化物10-30min;通入氧气10min,加入二甲硫醚(1.0mol),25-30℃反应2h;反应用二氯甲烷萃取,有机相用无水硫酸钠干燥,浓缩得到式9化合物;
10、8)在反应釜加入式9化合物(1.0mol)、乙二醇(2.5-3.0mol)、dce、dmf的混合溶剂,在0℃~-5℃搅拌下滴加酸(1.0-1.1mol),滴加完毕后在80-85℃搅拌反应5h,反应液加入10%碳酸钠溶液洗涤,二氯甲烷萃取,有机相用无水硫酸钠干燥,浓缩得到式10化合物。
11、本发明优选实施方案:
12、步骤1)所需的氯代试剂可以选2-氯-1,3-二甲基氯化咪唑啉、二氯亚砜、三氯氧磷等,优选2-氯-1,3-二甲基氯化咪唑啉;
13、步骤2)所需的具有活性亚甲基的化合物可以选丙二酸亚异丙酯、丙二酸酯、β-酮酸酯,优选丙二酸亚异丙酯;所需的胺可以选三乙胺、1,8-二偶氮杂双螺环[5.4.0]十一-7-烯等,优选三乙胺;
14、步骤3)所需的胺可以选二甲基乙二胺、三乙胺等,优选三乙胺;
15、步骤4)所需乙基化试剂可以选碘乙烷、溴乙烷等,优选碘乙烷;
16、步骤5)水解所需的酸可以选浓硫酸、浓盐酸等,优选浓硫酸;
17、步骤6)催化剂可以选四丁基溴化铵、三乙基苄基溴化铵等,优选三乙基苄基溴化铵;脱羧用的酸可以选氢溴酸、盐酸、硫酸等,优选氢溴酸;
18、步骤7)过氧化物可以选过氧化氢、高锰酸钾、臭氧等,优选臭氧。
19、步骤8)乙二醇保护所需的酸可以选对甲苯磺酸、三甲基氯硅烷等。优选三甲基氯硅烷。
20、本发明典型的反应线路如下:
21、
22、本发明的有益效果在于:涉及的反应多为常规反应,反应条件较为温和,相较于前人工艺,降低了反应的复杂度与降低了安全性;用到的物料较为廉价且工艺过程收率提高,使得总体成本降低;且合成路线短,产率较高,过程易于纯化。
- 还没有人留言评论。精彩留言会获得点赞!