一种巴瑞替尼及其中间体的新合成方法与流程
本发明属于医药,具体涉及一种选择性口服jak1/jak2抑制剂药物巴瑞替尼的中间体及其合成方法和用该中间体制备巴瑞替尼的方法。
背景技术:
1、巴瑞替尼(baricitinib)是由eli lilly公司与incyte公司合作开发的选择性口服jak1/jak2抑制药,能抑制白细胞介素-6(il-6)和白细胞介素-23(il-23)等多种炎性细胞因子的细胞内信号传导,可用于中度至重度类风湿性关节炎(ra)的治疗。2017年2月,巴瑞替尼获欧盟批准上市,获批适应症为类风湿关节炎,这是欧盟批准的治疗类风湿关节炎的首个jak抑制剂药物。2018年6月获得fda批准被用于治疗某些患有中度至重度活动性类风湿关节炎的成年患者,2019年被批准在中国上市,2020年12月纳入医保。
2、2022年6月13日,fda批准巴瑞替尼口服片剂用于治疗患有严重斑秃的成年患者,该疾病每年在美国影响超过300000人,此次获批是fda首次批准斑秃的全身疗法,巴瑞替尼作为jak抑制剂的优势品种,未来潜力巨大。
3、巴瑞替尼的化学名称为1-(乙基磺酰基)-3-[4-(7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基]-3-氮杂环丁烷乙腈,其结构式如下:
4、
5、目前,国内外报道的关于巴瑞替尼的合成方法有以下几种:
6、1、incyte公司在专利wo 2009114512/cn 102026999b公开的巴瑞替尼及其关键中间体的合成路线。
7、
8、该路线以二苯甲胺为原料,依次经与氯甲基环氧乙烷反应、高压催化氢化脱二甲苯基保护基、氨基保护、氧化反应、wittig反应、脱boc保护及磺酰胺化6步反应得到2-[1-(乙基磺酰基)氮杂环丁烷-3-亚甲基]-乙腈20。4-氯吡咯并嘧啶(11)经三甲基硅烷基乙氧甲基氯(semcl)进行氨基保护后,与1-(1-乙氧基乙基)-4-吡唑基硼酸频哪醇酯进行suzuki偶联,然后经盐酸脱1-乙氧基乙基保护制得吡唑化合物24。中间体24与中间体20在dbu催化下经迈克尔加成反应制备得到中间体25,经libf4和nh4oh两步脱保护得到最终产物巴瑞替尼。
9、该路线制备中间体2-[1-(乙基磺酰基)氮杂环丁烷-3-亚甲基]-乙腈20的步骤较长,使用的氰甲基膦酸二乙酯价格较高,导致该中间体的成本过高;合成吡唑中间体24的过程中使用三甲基硅烷基乙氧甲基氯保护时需使用到氢化钠,工艺操作危险性较大,而且脱去三甲基硅烷基乙氧甲基保护基时需要两步反应,效率较低,操作繁琐,总收率低,成本较高,不适合工业化生产。
10、2、专利cn105294699a公开的合成巴瑞替尼的方法。
11、
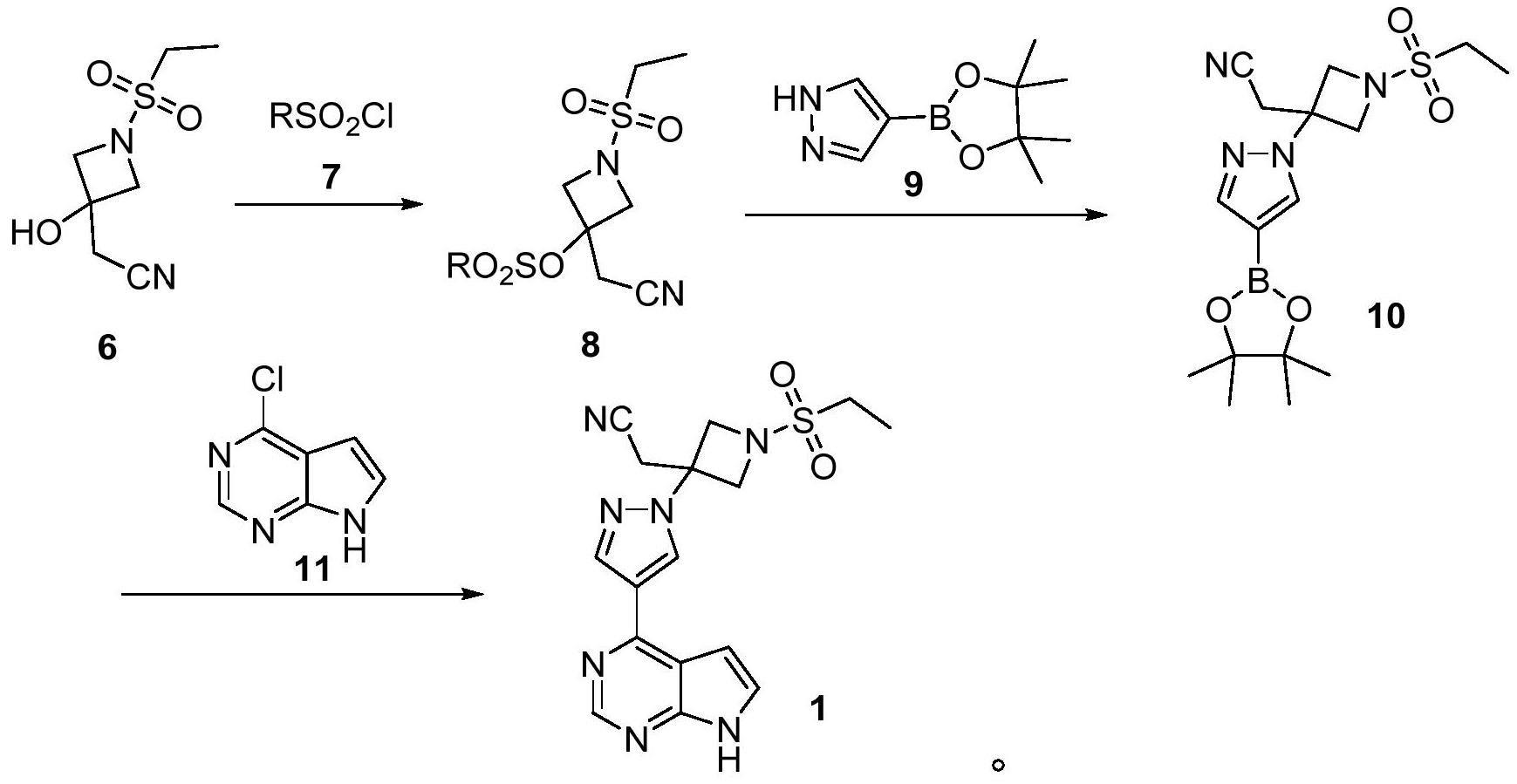
12、以4-吡唑硼酸频哪醇酯9为起始原料,与3-(氰基亚甲基)氮杂环丁烷-1-甲酸叔丁脂18经迈克尔加成反应制得中间体3-(氰基亚甲基)-3-(4-吡唑硼酸频哪醇酯)氮杂环丁烷-1-甲酸叔丁酯27。27与boc保护的4-氯-7-(1-甲酸叔丁酯)-7h-吡咯并[2,3-d]嘧啶28经钯催化偶联反应得双boc保护的化合物29,经脱两分子boc保护基制得3-(氰基亚甲基)-3-((7h-吡咯并[2,3-d]嘧啶-4-基)-1h-吡唑-1-基)氮杂环丁烷30。最后,再与乙基磺酰氯在发生磺酰化反应得到终产物巴瑞替尼。
13、该方法使用的原料3-(氰基亚甲基)氮杂环丁烷-1-甲酸叔丁脂18不易得,且脱去boc保护基后的中间体有两个活性氨基,最后一步与乙基磺酰氯在发生磺酰化反应时容易产生双取代的副产物或者更易进攻吡咯环上的氨基,导致副产物多,不易控制。
14、3、专利cn 106946917公开的一种制备巴瑞替尼及其中间体的新方法。
15、
16、该方法以1-氨基-3-氯丙-2-醇盐酸盐31为起始原料,在碱的作用下与乙磺酰氯发生反应对氨基进行保护,接着在碱的作用下环合,得到氮杂环丁烷33,被氧化得到氮杂环丁酮4,氮杂环丁酮4与三苯基膦乙腈发生wittig反应得到2-[1-(乙基磺酰基)氮杂环丁烷-3-亚甲基]-乙腈20,中间体20与硼酸酯化合物反应得到中间体36,经偶联反应和脱保护反应得到巴瑞替尼。该方法直接用乙磺酰基保护氨基后再利用碱的作用直接关环得到氮杂环丁烷中间体,避免使用其他保护基。但是该方法存在反应路线较长,部分原料制备困难,且wittig反应的副产物三苯基氧膦难以分离等问题。
技术实现思路
1、本发明要解决的技术问题是克服现有技术的不足,提供一种巴瑞替尼的中间体及其合成方法和用该中间体制备巴瑞替尼的方法。该方法工艺路线简单、原料易得、易于操作、成本低廉、适合工业化生产。
2、为解决上述技术问题,本发明采取如下技术方案:
3、本发明首先提供了一种巴瑞替尼中间体化合物6的合成方法,其包括如下步骤:
4、(1)氮杂环丁烷-3-酮盐酸盐2和乙基磺酰氯3在碱的作用下发生磺酰化反应得到化合物4;
5、(2)化合物4和卤代腈5在催化剂的作用下发生reformatsky反应得到化合物6。
6、其合成路线为:
7、
8、进一步地,所述步骤(1)中氮杂环丁烷-3-酮盐酸盐2和乙基磺酰氯3的摩尔比为1:1-1.5。
9、进一步地,所述步骤(1)中的碱为三乙胺、n,n-二异丙基乙胺、碳酸钠或碳酸钾中的一种;溶剂为二氯甲烷、乙腈、四氢呋喃中的一种。
10、进一步地,所述步骤(2)中的催化剂为金属催化剂锌或碘化钐。
11、进一步地,所述步骤(2)中化合物4、卤代腈5和催化剂的摩尔比为1:1~2:1~2;反应溶剂为四氢呋喃、甲苯中的一种;反应温度为0~80℃。
12、本发明还提供了一种巴瑞替尼中间体化合物6,其结构式为:
13、
14、本发明还提供了由巴瑞替尼中间体化合物6合成巴瑞替尼的方法,包括如下步骤:
15、(1)化合物6和磺酰氯7在碱的作用下发生磺酰化反应得到化合物8;
16、(2)化合物8和化合物9发生缩合反应得到化合物10;
17、(3)化合物10与4-氯吡咯并嘧啶11进行suzuki偶联反应,得到化合物1,即巴瑞替尼。
18、
19、进一步地,所述步骤(1)中的磺酰氯为甲基磺酰氯、对甲苯磺酰氯、苯磺酰氯、对硝基苯磺酰氯中的一种;化合物6和磺酰氯的摩尔比为1:1~1.5。
20、进一步地,所述步骤(1)中的碱为三乙胺、三乙烯二胺(dabco)、氢氧化钠中的一种。
21、进一步地,所述步骤(2)中化合物8和化合物9的摩尔比为1:1~2。
22、进一步地,所述步骤(2)中催化剂为碳酸铯或碳酸钾;溶剂为n,n-二甲基甲酰胺或n,n-二甲基乙酰胺;反应温度为0~100℃。
23、进一步地,所述步骤(3)中suzuki偶联反应是在钯催化剂和碱的作用下进行的,所述钯催化剂为pd(pph3)4、pd(pph3)2cl2、pd(oac)2/pph3中的一种;碱为碱金属碳酸盐,优选碳酸钾。
24、基于以上对本发明的阐述,本发明还进一步提供了一种巴瑞替尼的合成方法,其包括如下步骤:
25、(1)氮杂环丁烷-3-酮盐酸盐2和乙基磺酰氯3在碱的作用下发生磺酰化反应得到化合物4;
26、(2)化合物4和卤代腈5在催化剂的作用下发生reformatsky反应得到化合物6;
27、(3)化合物6和磺酰氯7在碱的作用下发生磺酰化反应得到化合物8;
28、(4)化合物8和化合物9发生缩合反应得到化合物10;
29、(5)化合物10与4-氯吡咯并嘧啶11进行suzuki偶联反应,得到化合物1,即巴瑞替尼。
30、
31、与现有技术相比,本发明具有以下显著优点:
32、(1)本发明提供了一种新的巴瑞替尼中间体化合物6,该中间体可以用于巴瑞替尼的合成。该中间体以氮杂环丁烷-3-酮盐酸盐为起始原料,经乙基磺酰氯保护氨基后与卤代腈发生reformatsky反应得到。与现有的合成巴瑞替尼的中间体20相比,化合物6的合成步骤少,起始物料价格低廉、易得,不经过wittig反应,提高了路线效率和原子经济性,避免了现有中间体合成技术中的步骤长、原料价格昂贵、副产物三苯基氧膦分离困难等问题。
33、(2)本发明提供了由化合物6制备巴瑞替尼的方法,化合物6经磺酰化反应、缩合反应和偶联反应得到巴瑞替尼。该方法不使用危险性高的试剂(氢化钠、格式试剂等),反应条件温和;使用磺酰氯与羟基形成磺酰氧基,增加离去能力,提高了反应收率(羟基的离去能力是比较弱的,中间体6与9直接反应的收率很低);4-氯吡咯并嘧啶不需要进行氨基保护和脱保护,简化了反应工序,提高了整体反应效率。
34、(3)与现有技术相比,本发明提供的方法,后处理简单,综合成本低廉,没有危险工艺和特殊设备要求,适合工业化生产。
- 还没有人留言评论。精彩留言会获得点赞!