一种EDOT类化合物及其制备方法与在制备电致变色材料中的应用
(一)本发明涉及一种edot类化合物及其制备方法与在制备电致变色材料中的应用。
背景技术:
0、(二)背景技术
1、电致变色材料是指在外加电压的作用下发生可逆的颜色变化的一类新型功能材料,由于其环境友好且能耗低等优点被认为是新一代显示材料。但由于变色材料本身不发光,对于非发射显示器必须使用原色减色法(cmy)来产生各种颜色。
2、而现有技术中尚未出现基于cmy三原色实现全色显示的材料,这限制了电致变色材料的应用。
3、基于此,本发明设计并合成了三种单体:d-a-d型单体、d-π-d-π-d型单体、纯d型单体并通过循环伏安法制备了在中性态为青色、品红和黄色,氧化时呈现高透的高性能电致变色薄膜,并期望在未来的发展中进一步扩展其应用。
技术实现思路
0、(三)
技术实现要素:
1、本发明目的是提供一种edot类化合物及其制备方法与在制备电致变色材料中的应用,本发明提供了d、a、π结构的edot类化合物,制备的电致变色材料分别实现中性态下呈现青、品红、黄三种颜色,氧化时呈现高透明色,在三基色电致变色薄膜的基础上实现全色显示的目的。
2、本发明采用的技术方案是:
3、本发明提供一种edot类化合物,所述edot化合物为下列之一:
4、
5、
6、式(i)所示化合物为d-a-d型单体;式(ii)所示化合物为d-π-d-π-d型单体;式(iii)所示化合物为纯d型单体。
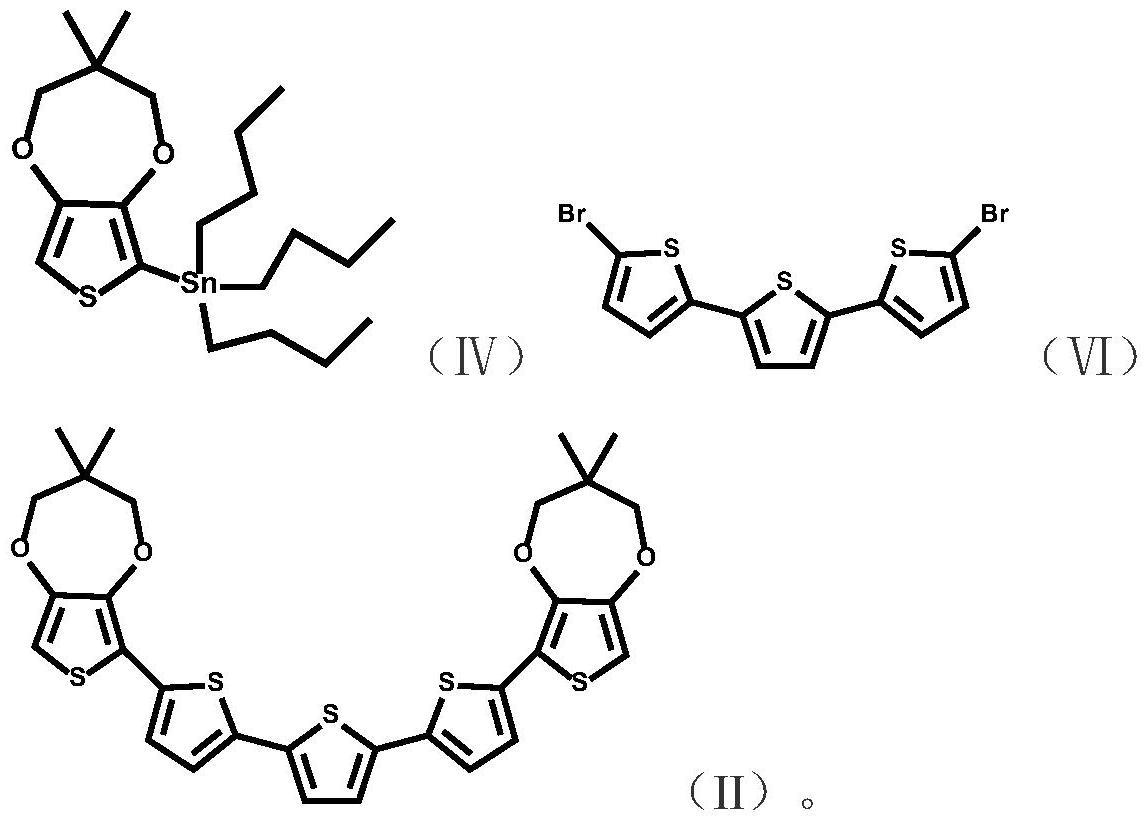
7、本发明还提供一种所述edot化合物的制备方法,所述化合物(i)按如下方法制备:在氮气保护下,式(ⅳ)所示化合物(即三丁基(3,3-二甲基-3,4-二氢-2h-噻吩并[3,4-b][1,4]二氧六环-6-基)锡烷)与式(ⅴ)所示4,7-二溴苯并噻二唑,在四(三苯基膦)钯(pd(pph3)4)、超干n,n-二甲基甲酰胺(dmf)作用下,回流反应完全后,反应液后处理,得到式(i)所示化合物,记为bt。所述式(ⅳ)所示化合物与式(ⅴ)所示4,7-二溴苯并噻二唑的投料物质的量之比为2~4:1(优选3:1);所述超干dmf的加入体积以4,7-二溴苯并噻二唑的质量计为50~100ml/g(优选70ml/g);所述pd(pph3)4与式(ⅳ)所示化合物和4,7-二溴苯并噻二唑总的物质的量之比为0.001-0.1:1,优选0.03:1;
8、
9、所述反应液后处理方法为:反应液冷却至室温,用二氯甲烷萃取反应混合物,有机相用饱和食盐水清洗三遍后,用无水na2so4干燥,采用200~300目硅胶在真空下浓缩拌样至完全去除溶剂,上样层析柱,所述层析柱的上层为石英砂,底层为300-400目硅胶,以体积比1:2的二氯甲烷/石油醚为洗脱剂,自然洗脱,收集全部流出液,减压浓缩至干,得到式(i)所示化合物。
10、本发明所述化合物(ii)按如下方法制备:在氮气保护下,式(ⅳ)所示化合物(即三丁基(3,3-二甲基-3,4-二氢-2h-噻吩并[3,4-b][1,4]二氧六环-6-基)锡烷)与式(ⅵ)所示化合物(即5,5'-二溴-2,2”:5',2”-三联噻吩),在pd(pph3)4、超干dmf作用下,回流反应完全,反应液分离纯化,获得式(ii)所示化合物,记为q3tq;所述式(ⅳ)所示化合物与式(ⅵ)所示化合物的投料物质的量之比为2~4:1(优选3:1);所述超干dmf的加入体积以式(ⅵ)所示化合物的质量计为50~100ml/g(优选70ml/g);所述pd(pph3)4与式(ⅵ)所示化合物和式(ⅵ)所示化合物总物质的量之比为0.001-0.1:1,优选0.03:1;
11、
12、所述反应液分离纯化的方法为:反应液冷却至室温,用二氯甲烷萃取反应混合物,有机相用饱和食盐水清洗三遍后,无水na2so4干燥,加入200~300目硅胶,在真空下浓缩拌样至去除溶剂,上样层析柱,所述层析柱的上层为石英砂,底层为300-400目硅胶,以体积比1:1的二氯甲烷/石油醚为洗脱剂,自然洗脱,收集全部流出液,减压浓缩至干,得到式(ii)所示化合物。
13、本发明所述化合物(iii)按如下步骤制备:
14、(1)氮气保护下,式(ⅶ)所示3,4-二甲氧基噻吩与2,2-双(溴甲基)丙烷-1,3-二醇,在对甲苯磺酸、超干甲苯作用下,回流生成式(ⅷ)所示化合物(即3,3-双(溴甲基)-3,4-二氢-2h-噻吩并[3,4-b][1,4]二氧杂环丁烷);所述2,2-双(溴甲基)丙烷-1,3-二醇与3,4-二甲氧基噻吩的投料物质的量之比为1~2:1(优选1.1:1);所述对甲苯磺酸与3,4-二甲氧基噻吩的投料物质的量之比为0.1~0.3:1(优选0.1:1);所述超干甲苯的加入体积以3,4-二甲氧基噻吩的质量计为80~130ml/g(优选100ml/g);
15、(2)氮气保护下,式(ⅷ)所示化合物与2-乙基己醇,在氢化钠和超干dmf作用下,回流反应生成式(ⅸ)所示化合物(即3,3-双-(2-乙基己氧基氧基)甲基)-3,4-二氢-2h-噻吩并[3,4-b][1,4]二氧杂环丁烷);所述氢化钠与式(ⅷ)所示化合物的投料物质的量之比为0.4~0.7:1(优选0.5:1);所述2-乙基己醇与式(ⅷ)所示化合物的投料物质的量之比为0.3~0.5:1(优选0.3:1);所述超干dmf的加入体积以式(ⅷ)所示化合物的质量计为1.8~3ml/g(优选1.9ml/g);
16、(3)在-78℃、氮气保护的条件下,将式(ⅸ)所示化合物、超干四氢呋喃(即无水无氧四氢呋喃)加入到烧瓶中,向其中逐滴滴加n-丁基锂(n-buli)的正己烷溶液,保温搅拌30-45min后,向其中加入三丁基氯化锡,反应生成式(ⅹ)所示化合物(即三丁基锡化3,3-双-(2-乙基己氧基氧基)甲基)-3,4-二氢-2h-噻吩并[3,4-b][1,4]二氧杂环丁烷);所述超干四氢呋喃的加入体积以式(ⅸ)所示化合物的质量计为10~20ml/g(优选14ml/g);所述n-buli的正己烷溶液浓度为1.6-2.4m,所述n-buli与式(ⅸ)所示化合物的投料物质的量之比为1~1.5:1(优选1.05:1);所述三丁基氯化锡与式(ⅸ)所示化合物的投料物质的量之比为1~1.5:1(优选1.1:1);
17、(4)氮气保护下,式(ⅺ)所示5,5-二溴-3,4-乙烯二氧噻吩与苯硼酸,在pd(pph3)4、超干四氢呋喃作用下,于k2co3水溶液中,反应生成式(ⅻ)所示2,5-二苯基-3,4-乙烯二氧噻吩;所述苯硼酸与5,5-二溴-3,4-乙烯二氧噻吩的投料物质的量之比为2~2.3:1;所述超干四氢呋喃的加入体积以5,5-二溴-3,4-乙烯二氧噻吩的质量计为2~2.5ml/g;所述k2co3水溶液浓度为2m,加入体积以5,5-二溴-3,4-乙烯二氧噻吩的质量计为1.4~1.6ml/g;所述pd(pph3)4与5,5-二溴-3,4-乙烯二氧噻吩和苯硼酸总物质的量之比为0.01-0.1:1,优选0.03:1;
18、(5)在0℃条件下,式(ⅻ)所示2,5-二苯基-3,4-乙烯二氧噻吩与氯仿稀释的液溴,在氯仿中反应生成式(i)所示化合物(即2,5-双(4’-溴苯基)-3,4-乙撑二氧噻吩);所述氯仿的加入体积以2,5-二苯基-3,4-乙烯二氧噻吩的质量计为4~8ml/g;所述稀释液溴的氯仿的加入体积以2,5-二苯基-3,4-乙烯二氧噻吩的质量计为18~25ml/g;所述液溴与2,5-二苯基-3,4-乙烯二氧噻吩的投料物质的量之比为2~2.5:1;
19、(6)在氮气保护下,式(i)所示化合物与式(ⅹ)所示化合物,在pd(pph3)4、超干dmf作用下反应生成式(ⅲ)所示化合物,记为probebpro;所述式(ⅹ)所示化合物与式(i)所示化合物的投料物质的量之比为2~4:1,优选3:1;所述超干dmf的加入体积以式(i)所示化合物的质量计为40~100ml/g,优选44ml/g;所述pd(pph3)4与式(i)所示化合物和式(ⅹ)所示化合物总物质的量之比为0.01-0.1:1,优选0.03:1;
20、
21、
22、优选的,步骤(1)按如下步骤进行:在氮气保护的条件下,将3,4-二甲氧基噻吩,2,2-双(溴甲基)丙烷-1,3-二醇、对甲苯磺酸、超干甲苯加入到烧瓶中,加热回流保持18h,冷却至室温,用二氯甲烷萃取,有机相用饱和食盐水清洗三遍后,用无水na2so4干燥,加入200~300目硅胶,在真空下浓缩拌样,上样层析柱,所述层析柱的上层为石英砂,底层为300-400目硅胶;以体积比1:3的二氯甲烷/石油醚为洗脱剂,自然洗脱,收集全部流出液,减压浓缩至干,得到式(ⅷ)所示3,3-双(溴甲基)-3,4-二氢-2h-噻吩并[3,4-b][1,4]二氧杂环丁烷。
23、优选的,步骤(2)按如下步骤进行:在氮气保护的条件下,将氢化钠加入烧瓶中、向其中滴加2-乙基己醇,常温搅拌30min后加入超干dmf,加热回流保持2.5h,再向加入3,3-双(溴甲基)-3,4-二氢-2h-噻吩并[3,4-b][1,4]二氧杂环丁烷,继续反应24h,冷却至室温,用二氯甲烷萃取反应混合物,有机相用饱和食盐水清洗三遍,用无水na2so4干燥,加入200~300目硅胶,在真空下浓缩拌样,上样层析柱,所述层析柱的上层为石英砂,底层为300-400目硅胶;以体积比2:3的二氯甲烷/石油醚为洗脱剂,自然洗脱,收集全部流出液,减压浓缩至无液体流出,得到目标产物3,3-双-(2-乙基己氧基氧基)甲基)-3,4-二氢-2h-噻吩并[3,4-b][1,4]二氧杂环丁烷(ⅸ)。
24、优选的,步骤(3)按如下步骤进行:在-78℃、氮气保护的条件下,将式(ⅸ)所示化合物、超干四氢呋喃(即无水无氧的四氢呋喃)加入到烧瓶中,向其中逐滴加入n-buli的正己烷溶液;保温搅拌30-45min后,向其中加入三丁基氯化锡,将反应混合物自然升至室温,搅拌过夜,上样以无水氧化铝为固定相的分离柱,以二氯甲烷为洗脱剂,快速洗脱,收集全部流出液,旋干除去溶剂后,得到式(ⅹ)所示化合物。
25、优选的,步骤(4)按如下步骤进行:在氮气保护的条件下,将5,5-二溴-3,4-乙烯二氧噻吩、苯硼酸、pd(pph3)4、超干四氢呋喃、k2co3水溶液加入到烧瓶中,加热回流保持18h,冷却至室温,用二氯甲烷萃取反应混合物,有机相用饱和食盐水清洗三遍,用无水na2so4干燥,旋干;粗产物用甲醇重结晶,得到目标产物2,5-二苯基-3,4-乙烯二氧噻吩(ⅻ)。
26、优选的,步骤(5)按如下步骤进行:在0℃的条件下,将2,5-二苯基-3,4-乙烯二氧噻吩、氯仿加入烧瓶中,向其中逐滴加入氯仿稀释后的液溴,室温下搅拌20-40min后,依次用质量浓度3% naoh水溶液,饱和nahso3水溶液和去离子水洗涤,有机层用无水na2so4干燥,旋干;粗产物用石油醚重结晶,得到2,5-双(4’-溴苯基)-3,4-乙撑二氧噻吩(i)。
27、优选的,步骤(6)按如下步骤进行:在氮气保护的条件下,将式(i)所示化合物、式(ⅹ)所示化合物、pd(pph3)4、超干dmf加入烧瓶中,加热回流保持18h,冷却至室温,用二氯甲烷萃取反应混合物,有机相用饱和食盐水清洗三遍,用无水na2so4干燥,加入200~300目硅胶,在真空下浓缩拌样去除溶剂上样层析柱,所述层析柱的上层为石英砂,底层为300-400目硅胶;以体积比1:1的二氯甲烷/石油醚为洗脱剂,自然洗脱,收集全部流出液,减压浓缩至无液体流出,得到式(ⅲ)所示化合物。
28、本发明还提供一种所述edot类化合物在制备透明电致变色材料中的应用,所述式(i)所示化合物用于制备青色-透明电致变色薄膜,式(ii)所示化合物用于制备品红-透明电致变色薄膜,式(ⅲ)所示化合物用于制备黄色-透明电致变色薄膜。
29、优选的,所述应用的方法为:将式(i)、式(ii)或式(ⅲ)所示化合物溶于含四丁基六氟磷酸铵(tbapf6)的二氯甲烷和乙腈的混合溶剂中,50khz超声5min,完全溶解之后,进行电化学聚合;以ito玻璃(0.9*4cm)为工作电极、铂丝为对电极、ag/agcl电极为参比电极,采用循环伏安法聚合成膜,电压范围为-0.3~1.2v(优选0~1.1v),扫描速度100mv/s,电聚合结束后,将覆盖有聚合物薄膜的ito玻璃在二氯甲烷和乙腈的混合清洗剂中清洗以除去薄膜表面未发生聚合的单体或低聚物以及残留的电解质,然后在空气中自然干燥,获得表面覆盖透明电致变色薄膜的ito玻璃;所述式(i)、式(ii)或式(ⅲ)所示化合物的浓度均为0.01~1.5mmol/l(优选0.01mmol/l),所述四丁基六氟磷酸铵的浓度为0.1~0.12mol/l(优选0.1mol/l);所述混合溶剂与混合清洗剂中二氯甲烷和乙腈的体积比均为2-4∶1。其中,式(i)所示化合物对应的二氯甲烷和乙腈的体积比均为4∶1;式(ii)或式(ⅲ)所示化合物对应的二氯甲烷和乙腈的体积比均为7∶3。
30、与现有技术相比,本发明有益效果主要体现在:本发明提供的edot类化合物分别可通过循环伏安法制备高性能青色-透明(pbt)、品红-透明(pq3tq)、黄色-透明(pprobebpro)电致变色薄膜。其中青色-透明(pbt)电致变色薄膜有着较低的变色电压(<0.7v),在770nm处,着色时间为0.63s,褪色时间为0.49s,经历600个循环后仍能保留初始值98%的光学对比度;品红-透明(pq3tq)电致变色薄膜有着较低的变色电压(<1v),在531nm处,着色时间为0.63s,褪色时间为0.29s,经历600个循环后仍能保留初始值96.9%的光学对比度;黄色-透明(p(probebpro))电致变色薄膜有着较低的变色电压(<1v),在444nm处,着色时间为1.58s,褪色时间为0.59s,经历600个循环后仍能保留初始值79.2%的光学对比度。以上这三种单体不仅丰富了电化学聚合制备聚合物的颜色库,更为后续电致变色聚合物颜色的调控提供了理论依据。
- 还没有人留言评论。精彩留言会获得点赞!