一种含过渡金属配离子的Mo-As-Se三元硒化物及其溶剂热制备方法与应用
本发明涉及硒化物材料,尤其是指一种含过渡金属配离子的mo-as-se三元硒化物及其溶剂热制备方法与应用。
背景技术:
1、过渡金属具有未充满的(n-1)d电子层结构,表现出良好的磁性和发光性能,而p区元素锑(sb)和砷(as)的多元金属硫属化合物具有良好的半导体性质,因此,含过渡金属的sb和as的多元金属硫属化合物是性能优良的多功能复合材料,而且其能隙可以通过选择不同的过渡金属进行调节,在磁性材料、半导体材料和发光及催化复合材料等领域具有广泛的应用前景。因此,含过渡金属的sb和as多元金属硫属化合物的研究得到了人们的广泛关注。例如,含过渡金属离子的fe-sb-s三元锑硫化物[fe(tren)]fesbs4(tren=三(2-氨基乙基)胺)、[fe(dien)2]fe2sb4s10(dien=二乙烯三胺)和[fe(en)2]sb4s7(en=乙二胺),以及mn-sb-s三元硫化物(dienh3)[(dienh)mnsb8s15]·h2o是性能良好的磁性材料;ag-sb-s三元硫化物[nh4]agsb4s7·h2o和[nh4]agsb2s4对光降解结晶紫均具有很强的催化作用;cu-sb-se三元硒化物cusbse3·0.5en和cusbse3·en是潜在的具有窄能隙的半导体材料,其能隙分别为1.58ev和1.61ev。mn-as-s三元砷硫化物[nh4]mnas3s6是具有弱反铁磁性的磁性材料,而{[mn(phen)3](ass3)2}·h2o(phen=1,10-菲啰啉)的磁性具有自旋斜交控制特性。mn-as-se三元硒化物[mn(phen)2]as2se3(se2)的半导体能隙为2ev,并具有反铁磁性相互作用。
2、钼(mo)和砷(as)是典型的亲硫元素,可以和硫属元素形成结构多样、组成稳定的mo-e和as-e(e=o,s,se)二元硫属化合物。目前,人们已经合成和表征了一系列的mo-s和as-s二元硫化物,例如,含有机阳离子的钼硫化合物[(ch3)4n]2[mo2o2(μ-s)2(s2)2]、[(ch3)4n]2[mo2o2(μ-s)2(s2)(s4)]、[(ch3)4n]2[mo2o2(μ-s)2(s2)(s4)]·ch3cn和[ph4p]2[mo2o2(μ-s)2(s2)2],以及含有机配体的钼硫化合物[mo2o2(μ-s)2(dmf)3][17]、mo2o2(μ-s)2(et2dtc)2(dtc=dithiocarbamate)。as-s二元硫化物有tl3ass3、[mn(dien)2]n[mn(dien)ass4]2n·4nh2o、[mn(en)3]2[mn(en)2ass4][as3s6]、[mn3(2,2'-bipy)3(ass4)2]n·nh2o(2,2'-bipy=2,2'-联吡啶)和[mn3(phen)3(ass4)2]n·nh2o等。mo-se和as-se二元硒化物的研究也有一定的文献报道,但是,未见含过渡金属配离子的mo-as-se三元硒化物的报道。
3、一.关于mo-se二元硒化物的报道
4、(1)1990年,henkel分别以(nh4)6mo7o24·6h2o和nahse作为钼源和硒源,以cys(cys=l(+)-半胱氨酸)为有机配体,制备了钼硒化合物na[(ch3)4n][mo2o2(μ-se)2(cys)2]·7h2o,由于经多步反应合成,产率较低,仅为33%。(参见:g.henkel,g.kampmann,b.krebs,g.j.lamprecht,m.nasreldin,a.g.sykes,j.chem.soc.,chem.commun.,1990,15,1014-1016.)。
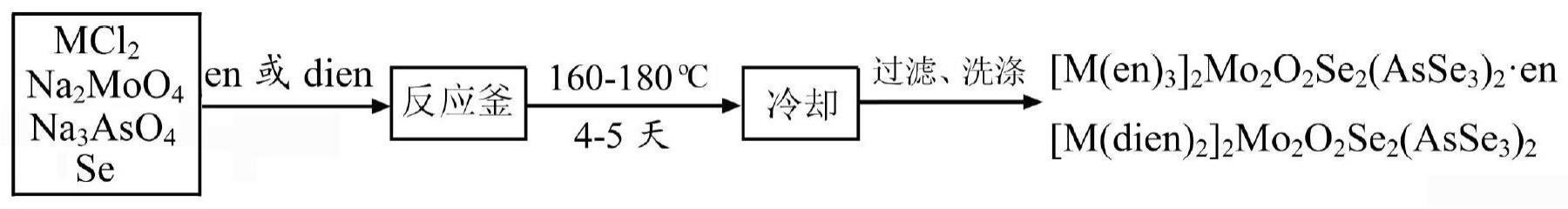
5、(2)1992年,kanatzidis用mo、k2se4、h2o按照1:1.5:22.2的摩尔比于135℃反应3天,冷却至室温后用乙醇洗涤,得到钼硒化合物k2mo3se18,产率为20%;moo3、k2se4、h2o按照1:2:22.2的摩尔比在相同温度下反应3天,产物经乙醇洗涤,得到钼硒化合物k8mo9se40·4h2o,产率为约20%。1995年,kanatzidis用moo3、k2se4、me4ncl按照1:3:2的摩尔比于135℃反应3天,冷却至室温后用dmf洗涤除去sex2-和nacl杂质,得到了含有机阳离子的钼硒化合物(me4n)2mo3se13。上述合成的操作过程均在手套箱中于氮气气氛中进行。(参见:j.liao,m.kanatzidis,inorg.chem.,1992,31,431-439;inorg.chem.,1995,34,2658-2670)。
6、(3)1993年,eichhorn首先用mo2(o2cch3)4(o2cch3为乙酸根)和k2se4在乙二胺中反应3小时,过滤得到清液,然后向滤液加入冠醚2,2,2-crypt(2,2,2-crypt=1,4,7,10,13,16-六氧杂环十八烷),反应完毕后过滤,滤液静置6天后得黑色晶体[k(2,2,2-crypt)]2[mo2o2(μ-se)2(se2)],产率为55%。合成原料k2se4是在氮气气氛保护下,由钾和硒在液氨中反应制备的。合成中使用的dmso、dmf溶剂要不含水分,使用前分子筛干燥后进行蒸馏。(参见:b.w.eichborn,j.r.gardner,a.n.ziebah,k.j.ahmeti,s.g.bott,inorg.chem.,1993,32,5412-5414.)。
7、(4)1996年,mak首先用mo(co)6和k2se4在乙醇中于100℃反应72小时,得到黑色粉末,然后用黑色粉末与et4ncl·h2o在乙醇中反应,制备了钼硒化合物(et4n)2[(mo2o2se6)0.20((mo2o2se6)0.18(mo2o2se6)0.62]。1997年,mak把mo(co)6、et4ncl·h2o、ag、k2se4混合在乙醇中,把混合物转入玻璃管,用液氨冷却后把玻璃管密封,于100℃反应72小时,然后以每小时降低6℃的速度冷却到50℃,得到钼硒化合物[et4n]6mo6se42,产率为5%。该化合物的制备需两步反应,产率较低,要使用剧毒性的mo(co)6作为钼源。且合成原料k2se4是在苛刻条件下制备,其制备方法是:在氮气气氛保护下,用化学计量比的钾和硒单质在高压釜中于400℃反应5小时得到k2se4。(参见:g.c.guo,t.c.w.mak,inorg.chem.,1998,37,6538-6540;g.c.guo,t.c.w.mak,dalton trans.1997,709–710.)。
8、(5)2006年,nguyen用mocl5和(ipr2pse)2se(ipr=异丙基)在乙醇中反应1小时,生成黄色沉淀,用乙醇洗涤沉淀后,在二氯甲烷中重结晶,得到棕色晶体[mo2o2(μ-se)2(se2pipr2)2]。硒源(ipr2pse)2se是由net3、ipr2pcl、hsicl3、se为原料,经过在己烷溶剂和甲苯溶剂中的两步反应,最后在甲苯中重结晶而合成的。[mo2o2(μ-se)2(ipr2pse2)2]和(ipr2pse)2se的合成均需要惰性环境,全程在标准schlenk无水无氧双排管操作系统进行。(参见:c.q.nguyen,a.adeogun,m.afzaal,m.a.malik,p.o.brien,chem.commun.,2006,20,2182-2184;c.q.nguyen,a.adeogun,m.afzaal,m.a.malik,p.o.brien,chem.commun.,2006,20,2179-2181.)。
9、(6)2022年,elliott利用多步反应法,在氮气保护下制备了含(ch3)4n+阳离子的钼硒化合物。一定量的硒粉加入水中,向水中通入氮气以排除水中的氧气(即脱气),然后加入nabh4,继续用氮气进行脱气,于50℃反应40分钟。然后,加入七钼酸铵(nh4)6mo7o24,在氮气流动下搅拌过夜后过滤。在滤液中加入四甲基氯化铵(ch3)4ncl,生成深棕色沉淀,分别用冷水、冷乙醇和乙醚洗涤沉淀。把沉淀溶解在dmf形成饱和溶液,饱和溶液静置数周后析出棕黑色晶体[(ch3)4n]2[mo2o2(μ-se)2(se2)2]。以moo3代替(nh4)6mo7o24,以se和na2se2代替硒粉,用上述相同的合成方法,最后在乙腈/乙醚混合溶剂中重结晶,制备了[(ch3)4n]2[mo2o2(μ-se)2(se2)(se4)]0.5[mo2o2(μ-se)2(se2)(se3)]0.25[mo2o2(μ-se)2(se2)]0.25。(参见:a.elliott,j.mcallister,l.masaityte,m.s.centellas,d.l.long,a.y.ganin,y.f.song,c.bo,h.n.miras,chem.commun.,2022,58,6906-6909.)。
10、二.关于as-se二元硒化物的报道
11、(1)1995年,kolis经多步反应制备了含co配体的砷硒化合物。把as4se4加入dmf,搅拌12小时后,加入fe(co)5,继续于100℃搅拌12小时,然后加入pph4br,再搅拌1小时,过滤后用蒸馏过的乙醚进行层析,层析后的溶液在4℃静置12小时,得到化合物(pph4)2[fe(as3se3)2(co)],产率为44%。用mn2(co)10代替fe(co)5,用相同的方法,得到混合(pph4)2[mn(as3se5)(co)3],产率为33%。上述实验过程全程在标准schlenk无水无氧(在氩气中)双排管操作系统进行。(参见:t.m.martin,p.t.wood,g.l.schimek,w.t.pennington,j.w.kolis,inorg.chem.,1995,34,4385–4391)。
12、(2)1998年,müller用as2se3、pph4br和na2se在ch3cn中于60℃反应24小时,过滤,滤液在-20℃结晶,得到含多硒离子se52-的砷硒化合物pph4[asse(se5)],提高ch3cn的用量,则得到砷硒化合物(pph4)2[as2se6]·2ch3cn,上述合成的操作过程需要在隔绝空气气氛中进行。(参见:j.w.czado,u.müller,z.anorg.allg.chem.,1998,624,239-243.)。
13、(3)1998年,ibers用asse4在乙二胺(en)中于60℃超声8小时,所得红色溶液用含有net4br的en溶液和甲苯进行层析,得到(net4)2[as2se6]和(net4)[asse8],由于经过多步反应和分离,产率较低,(net4)2[as2se6]和(net4)[asse8]产率分别为59%和22%。用tlasse4在乙二胺(en)中于60℃超声12小时,加入含有冠醚2,2,2-crypt(2,2,2-crypt=1,4,7,10,13,16-六氧杂环十八烷)的en溶液,然后用甲苯进行层析,蒸发除去甲苯后加入ch3cn,继续用乙醚层析,得到(enh)[asse6]·2,2,2-crypt,产率为18%。实验原料asse4、tlasse4由as、tl和se在n2气气氛中于高温反应制得;实验中用到的en、ch3cn、甲苯、乙醚等溶剂要经过干燥处理,通过蒸馏除去水分。(参见:d.m.smith,m.a.pell,j.a.ibers,inorg.chem.,1998,37,2340–2343)。
14、三.关于mo-as-se三元硒化物的报道
15、(1)1991年,kolis把as4se4和(c4h9)4n]2[mose4]加入dmf,搅拌24小时后过滤,滤液经过减压蒸馏除去溶剂dmf,然后加入ch2cl2溶剂,用蒸馏过的乙醚进行层析,层析后的溶液在4℃静置12小时,得到mo-as-se三元硒化物含有机阳离子的[(c4h9)4n]2[moas2se10],产率为10%。实验原料as4se4由as和se在n2气气氛中于600℃超声16小时反应制得;上述实验过程全程在标准schlenk无水无氧(在氩气中)双排管操作系统进行。(参见:s.c.o′neal,w.t.pennington,j.w.kolis,j.am.chem.soc.1991,113,710-712.)。
16、(2)1992年,kolis把as4se4加入dmf中,搅拌4小时后过滤,向滤液中加入mo(co)6,搅拌1小时后,然后加入(c4h9)4nbr,加热至100℃并搅拌8小时后过滤,滤液经过减压蒸馏除去溶剂dmf,然后加入ch2cl2溶剂,用蒸馏过的乙醚进行层析,层析后的溶液在4℃静置12小时,得到含有机阳离子的mo-as-se三元硒化物[(c4h9)4n]2[mo(co)2(as3se3)2],产率为20%。把as4se4和k2se3加入dmf中,搅拌4小时后过滤,向滤液中加入mo(co)6和(c4h9)4nbr,加热至100℃并搅拌2小时后过滤,滤液经过减压蒸馏除去溶剂dmf,然后加入ch2cl2溶剂,用蒸馏过的乙醚进行层析,层析后的溶液在4℃静置12小时,得到含有机阳离子的mo-as-se三元硒化物[(c4h9)4n]2[mo(asse5)2],产率为40%。实验过程全程在标准schlenk无水无氧(在氩气中)双排管操作系统进行。实验原料as4se4由as和se在n2气气氛中于600℃超声16小时反应制得,k2se3由k和se在液氨中于-78℃反应3小时制得。(参见:s.c.o′neal,w.t.pennington,j.w.kolis,inorg.chem.,1992,31,888–894)。
17、综上所述,虽然人们已经合成和表征了不少的mo-se和as-se二元硒化物,但是,国际上仅有三个mo-as-se三元硒化物的报道,而且这三个mo-as-se三元硒化物的抗衡阳离子均为有机阳离子(c4h9)4n+,含过渡金属配离子的mo-as-se三元硒化物的国内外均未见文献报道。上述mo-as-se三元硒化物以及mo-se和as-se二元硒化物的制备都需要多步反应,实验原理需要在特殊条件下制备,需要用到大量的有机溶剂,并需要在无水无氧环境下进行。
18、目前,人们制备mo-as-se三元硒化物以及mo-se和as-se二元硒化物现有技术是:(1)需要无水、无氧操作系统,通常在氮气或者氩气保护下进行实验,以确保隔离空气。(2)需要先合成mo(co)6、(nh4)6mo7o24、mocl5、as4se4、k2se4、k2se3、na2se2、(ipr2pse)2se等化合物作为合成中间体,这些中间体分别作为合成目标产物的钼源、砷源和硒源,这些中间体的制备需要在无水无氧的惰性或者在高温等苛刻条件下进行,比如,mo(co)6、as4se4、k2se3、和(ipr2pse)2se的制备是在n2气保护下完成的,k2se4和na2se2的制备是在氮气气氛下于400℃反应5小时得到的;as4se4由as和se在n2气气氛中于400℃和600℃高温条件下超声16小时反应制得,增加了制备成本,制备k2se4、k2se3、na2se2时需要使用的钾和钠是易燃易爆品,as4se4和mo(co)6是剧毒物质,这些增加了实验不安全性。(3)制备需要多步反应,需要两步或两步以上的反应,包括原料制备、反复萃取、层析、沉淀、结晶和重结晶等反应或者操作步骤,由于合成步骤多,造成产物收率较低,比如[(c4h9)4n]2[moas2se10]产率为10%;[(c4h9)4n]2[mo(co)2(as3se3)2]产率为20%;[(c4h9)4n]2[mo(asse5)2],产率为40%;[et4n]6mo6se42产率仅为5%;(enh)[asse6]·2,2,2-crypt,产率为18%。(4)制备过程通常在dmf(n,n-二甲基亚酰胺)、ch3cn(乙腈)、乙醚、甲苯、二氯甲烷等挥发性有机溶剂中进行,需要用有机溶剂反复萃取、层析、沉淀、结晶和重结晶等多步操作过程,可见有机溶剂用量大,且这些溶剂在使用前通常要进行蒸馏等除水处理,消耗能源,也不利于环境保护。
19、目前,人们制备和研究的mo-as-se三元硒化物是以有机阳离子为抗衡阳离子,含过渡金属配离子的mo-as-se三元硒化物的研究未见文献报道,以一步反应制备mo-as-se三元硒化物的研究也未见文献报道。
技术实现思路
1、为解决上述技术问题,本发明公开了一种以乙烯多胺为溶剂和配体合成含过渡金属配离子的mo-as-se三元硒化物制备方法及其结构。
2、本发明的制备方法利用乙烯多胺的螯合配位作用,在溶剂热条件下经过一步反应合成含过渡金属配离子的mo-as-se三元硒化物,合成方法操作简单,分别用简单的钼酸钠(na2moo4)、砷酸钠(na3aso4)和硒粉作为钼源、砷源和硒源,避免了传统方法中需要在无水、无氧条件制备特殊的钼、砷和硒的化合物作为钼源、砷源和硒源的中间步骤,利用简单易得的原料,由一步反应合成含过渡金属配离子的mo-as-se三元硒化物,提高了产物收率。反应不需要在无水、无氧条件下进行,也不需要传统制备方法中所用到的大量有机溶剂,只需要用乙烯多胺(反应溶剂)和乙醇(洗涤溶剂)两种溶剂,溶剂也不需要做进一步的除水处理。
3、本发明的第一方面的目的是克服现有合成mo-as-se三元硒化物技术中存在的需要多步反应,造成最终产物收率较低的缺陷,多步反应和操作包括原料制备、反复萃取、层析、沉淀、结晶和重结晶等。
4、本发明的第二方面的目的是克服现有制备mo-as-se三元硒化物技术中存在的先要合成mo(co)6、as4se4、k2se4、k2se3、na2se2、(ipr2pse)2se等中间产物作为钼源、砷源和硒源的缺陷,而这些中间产物的合成需要在400℃(如k2se4)和600℃(如as4se4)以上高温、或者在复杂的无水、无氧操作系统中进行(如mo(co)6、(ipr2pse)2se),而且使用大量dmf(n,n-二甲基亚酰胺)、ch3cn(乙腈)、乙醚、甲苯、二氯甲烷等易挥发的有机溶剂。
5、为了解决上述技术问题,本发明通过以下方案实现:
6、本发明的第一个目的在于提供一种含过渡金属配离子的mo-as-se三元硒化物,所述mo-as-se三元硒化物结构通式为:[m(en)3]2mo2o2se2(asse3)2·en或[m(dien)2]2mo2o2se2(asse3)2;其中,m为mn、co或ni,en为乙二胺,dien为二乙烯三胺。结构式中[m(en)3]2+和[m(dien)2]2+中m2+配位数均为6,m2+离子与6个n原子配位,形成mn6配位八面体。[mo2o2se2(asse3)2]4–由一个[mo2o2(μ-se)2)]2+双钼核和两个砷硒离子asse33–构成。阳离子核[mo2o2(μ-se)2)]2+由两个moo通过两个μ-se2–桥基配体连接而成,每个μ-se2–配体同时与两个mo原子连接。两个asse33–离子以双齿螯合配体形式分别与阳离子核[mo2o2(μ-se)2)]2+中的两个mo原子配位,最终形成mo-as-se三元阴离子[mo2o2se2(asse3)2]4–。
7、本发明的第二个目的在于提供一种含过渡金属配离子的mo-as-se三元硒化物的制备方法,包括以下步骤:将含m的盐、钼源、砷源、硒源在乙烯多胺溶剂中混合,超声搅拌,加热反应,得到所述含过渡金属配离子的mo-as-se三元硒化物;所述含m的盐中m为mn、co或ni。
8、在本发明的一个实施例中,所述含m的盐选自mncl2、cocl2和nicl2中的一种或多种。
9、在本发明的一个实施例中,所述钼源选自na2moo4和/或moo3。
10、在本发明的一个实施例中,所述砷源选自na3aso4和/或as。
11、在本发明的一个实施例中,所述硒源选自se和/或na2se。
12、在本发明的一个实施例中,所述乙烯多胺溶剂选自乙二胺和/或二乙烯三胺。乙二胺(沸点:116.5℃)和二乙烯三胺(沸点:207℃)沸点高,不易挥发,不易燃,避免了环境污染,同时提高了制备过程的安全性。本发明中乙烯多胺既是反应溶剂,也是过渡金属配离子的螯合配位剂,极性大,溶解性强,避免金属硒化物mose2、as2se3、mse(m=过渡金属mn,co,ni)沉淀的形成,有利于钼硒簇合物的结晶,从而形成mo-as-se三元硒化物晶体,提高产物的纯度;同时乙烯多胺具有很强的螯合配位能力,很容易与过渡金属配离子形成配离子,进而与mo-as-se三元阴离子结合,形成含过渡金属配离子的mo-as-se三元硒化物,乙烯多胺与过渡金属配离子在原位反应形成过渡金属配阳离子,做为mo-as-se三元硒化物的抗衡离子,从而形成含过渡金属配离子的mo-as-se三元硒化物。且乙二胺和二乙烯三胺不改变mo-as-se三元阴离子的结构。
13、在本发明的一个实施例中,所述含m的盐、钼源、砷源、硒源的摩尔比为1:1:1:4-1:1:1:5。
14、在本发明的一个实施例中,所述加热反应的反应条件:反应温度为160-180℃,反应时间为4-5天。
15、在本发明的一个实施例中,还包括含过渡金属配离子的mo-as-se三元硒化物的提纯,步骤为:加热之后,冷却至室温,过滤取滤饼,并用洗涤剂洗涤滤饼,得到所述含过渡金属配离子的mo-as-se三元硒化物。
16、进一步的,所述洗涤剂选自乙醇。
17、本发明的第三个目的在于提供所述含过渡金属配离子的mo-as-se三元硒化物在催化光降解有机染料中的应用。
18、进一步的,所述有机染料包括且不限于亚甲基蓝(mb)、结晶紫(cv)、罗丹明b(rhb)、甲基橙(mo)等。
19、本发明的上述技术方案相比现有技术具有以下优点:
20、本发明的含过渡金属配离子的mo-as-se三元硒化物合成方法简单,利用溶剂热一锅煮的方法,省去传统合成方法中的原料制备、萃取、层析、蒸馏和重结晶等中间步骤,用廉价、无毒的na2moo4、na3aso4和se分别作为钼源、砷源和硒源,经一步溶剂热反应首次制得含过渡金属配离子的mo-as-se三元硒化物;不使用传统合成方法中的大量有机溶剂;产物分离提纯方便,产率高;mo-as-se三元硒化物结构明确、稳定;通过过渡金属配离子的不同可以调控mo-as-se三元硒化物半导体能隙。
21、(1)、本发明使用一步溶剂热反应直接合成含过渡金属配离子的mo-as-se三元硒化物,减少合成的中间步骤,与现有技术相比,提高了合成效率和产物产率,含过渡金属配离子的mo-as-se三元硒化物产率达67%以上。
22、(2)、用廉价和无毒的na2moo4、na3aso4和se作为钼源、砷源和硒源,不需要合成特定的钼、砷和硒的化合物作为中间体,降低了能源消耗.
23、(3)、不使用易挥、易燃的有机溶剂,避免了环境污染;不使用as4se4、mo(co)6和金属钾等剧毒和易燃易爆药品,提高了合成和制备mo-as-se三元硒化物实验安全性。
24、(4)、通过过渡金属配离子的不同可以调控mo-as-se三元硒化物半导体能隙和半导体性能。
25、(5)、不使用乙二胺(en)、二乙烯三胺(dien)和乙醇以外的任何有机溶剂,乙二胺、二乙烯三胺和乙醇在使用前不需要包括蒸馏除水在内的任何预处理。制备含过渡金属配离子的mo-as-se三元硒化物整个过程中不需要在无水无氧的惰性条下进行。
26、(6)、本发明含过渡金属配离子的mo-as-se三元硒化物对光降解有机染料的具有催化活性,对亚甲基蓝(mb)、结晶紫(cv)等有机染料催化降解百分率达到92%以上。
27、(7)、为设计、合成新型mo-as-se三元硒化物积累理论和实践经验。
- 还没有人留言评论。精彩留言会获得点赞!