一种细胞分裂抑制剂的制备方法
本发明属于药物合成领域,具体涉及一种细胞分裂抑制剂的制备方法。
背景技术:
1、近年来,二聚氨基酸类活性天然产物因其普遍具有的良好生物活性而受到药学届的广泛关注。以天然来源的ddm-a (didemnidine a)为代表的色氨酸-组氨酸异源二聚体在抑制细胞分裂g2期检查点等方面表现出了突出活性,同时其还具有一定的拓扑异构酶i和蛋白激酶c抑制作用,上述生物活性使其成为了抗癌、抗疟等药物的理想分子模型和成药前体。ddm-a的结构式如下:
2、。
3、目前,天然来源的ddm-a主要来自于海洋中的脊索动物——海鞘,ddm-a在自然界中的主要功能是对潜在的掠食者起到警示和防御的作用。近年来,由于海洋资源的过度开发和环境污染,造成ddm-a的自然来源的产量极其有限。
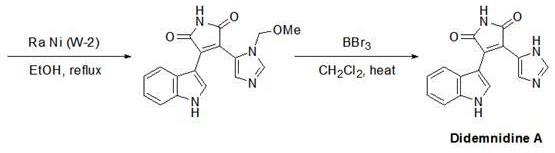
4、目前常用化学合成的方式制备ddm-a及其衍生物,其合成路线如下:
5、
6、
7、但上述技术方案存在以下不足之处:1、制备路线较长,方法为多步反应,较为复杂与繁琐,每步反应结束后均需要单独进行分离纯化;2、反应原料和反应副产物常具毒性,容易造成化学污染,生物安全性存疑;3.合成ddm-a的效率不高,整体回收产率普遍低于50%,且反应规模通常为百毫克级别,无法量产。
技术实现思路
1、发明目的:本发明针对上述现有技术存在的问题做出改进,即本发明公开了一种细胞分裂抑制剂的制备方法,其能够通过一锅两步法直接制备作为细胞分裂抑制剂的ddm-a。
2、技术方案:一种细胞分裂抑制剂的制备方法,具体步骤如下:
3、步骤一:关键合成酶的合成和载体构建
4、(11)合成用于表达关键合成酶的laad基因片段、viob基因片段、marc基因片段,其中:
5、所述laad基因片段的氨基酸序列如seq id no.1所示;
6、所述viob基因片段的氨基酸序列如seq id no.2所示;
7、所述marc基因片段的氨基酸序列如seq id no.3所示;
8、(12)利用gibson拼接克隆技术,将所述laad基因片段克隆到pet28a(+)载体中得到表达载体pet28a(+)-laad,将所述viob基因片段、marc基因片段分别克隆到pet26b载体中得到表达载体pet26b-viob、表达载体pet26b-marc,其中:
9、laad基因片段、viob基因片段、marc基因片段均位于ndei和hindiii限制性酶切位点之间;
10、(13)将构建好的表达载体pet28a(+)-laad、表达载体pet26b-viob、表达载体pet26b-marc 分别转化到感受态细胞bl21(de3)中得到菌种溶液,分别于-80℃保存;
11、步骤二:关键合成酶的表达和预处理
12、(21)制备laad蛋白粗制裂解液
13、(211)取10 ul的pet28a(+)-laad菌种溶液,加入到5 ml 的lb液体培养基中,37℃、200rpm震荡过夜培养,得到pet28a(+)-laad种子液,其中:
14、lb液体培养基中含卡那霉素,其浓度为50 ug/ml;
15、(212)将步骤(211)的得到的pet28a(+)-laad种子液全部加入到含1l的lb液体培养基的试验容器中,然后在摇床内以37℃、200rpm震荡培养,至菌液中含laad表达载体的浓度od600 = 1.1-1.3为止,随后将试验容器取出并置于冰浴中30 min,其中:
16、lb液体培养基中含卡那霉素,其浓度为50 ug/ml;
17、(213)向经过步骤(212)处理的菌液中加入适量的iptg,使得iptg在菌液中的浓度为0.1 mm,混匀后放入摇床内以18℃、200rpm诱导培养16 h;
18、(214)将经过步骤(213)诱导完毕的菌液倒入离心杯,4000g离心15 min,弃上清液,沉淀表面用ddh2o清洗,随后加入30 ml上样缓冲液将沉淀菌体重悬至无明显菌块得到菌体重悬液,其中:
19、所述上样缓冲液的ph值为8.0;
20、所述上样缓冲液包括:100 mm tris-hcl、300 mm nacl、5 mm咪唑、10%v/v 甘油,余量为水;
21、(215)利用超生破碎仪将步骤(214)得到的菌体重悬液破碎得到破碎后的菌液,其中:
22、变幅杆直径为6 mm;
23、破碎程序为:能量40%,超声循环工作时间2 s、暂停时间4 s,工作总时间30 min;
24、(216)将破碎后的菌液以10000g低温离心15min,取上清液,即得到laad蛋白粗制裂解液;
25、(22)制备viob蛋白粗制裂解液
26、(221)取10 ul的pet26b-viob菌种溶液,加入到5 ml的lb液体培养基中,37℃、200rpm震荡过夜培养,得到pet26b-viob种子液,其中:
27、lb液体培养基中含卡那霉素,其浓度为50 ug/ml;
28、(222)将步骤(221)得到的pet26b-viob种子液加入到含1l的lb液体培养基的试验容器中,在摇床内以37℃、200rpm震荡培养至含viob表达载体的浓度od600 = 0.7-0.9为止,随后将试验容器并置于冰浴中30 min,其中:
29、lb液体培养基中含卡那霉素,其浓度为50 ug/ml;
30、(223)向经过步骤(222)处理的菌液中分别加入适量的硫酸亚铁铵、5-氨基乙酰丙酸、iptg,使得硫酸亚铁铵在菌液中的浓度为40 um,5-氨基乙酰丙酸在菌液中的浓度为0.25 mm,iptg在菌液中的浓度为0.1 mm,然后混匀后放入摇床内以18℃、200rpm诱导培养16 h;
31、(224)将经过步骤(223)诱导完毕的菌液倒入离心杯,4000g离心15 min,弃上清液,沉淀表面用ddh2o清洗,随后加入30 ml上样缓冲液将沉淀菌体重悬至无明显菌块得到菌体重悬液,其中:
32、所述上样缓冲液的ph值为8.0;
33、所述上样缓冲液包括:100 mm tris-hcl、300 mm nacl、15 mm咪唑、10%v/v 甘油,余量为水;
34、(225)利用超生破碎仪将步骤(224)得到的菌体重悬液破碎,其中:
35、变幅杆直径为6 mm;
36、破碎程序为:能量40%,超声循环工作时间2 s、暂停时间4 s,工作总时间30 min;
37、(226)将破碎后的菌液以10000g低温离心15min,取上清液,即得到viob蛋白粗制裂解液。
38、(23)制备marc蛋白粗制裂解液
39、(231)取10 ul的pet26b-marc菌种溶液,加入到5 ml的lb液体培养基中,37℃、200rpm震荡过夜培养,得到pet26b-marc种子液,其中:
40、lb液体培养基中含卡那霉素,其浓度为50 ug/ml;
41、(232)将步骤(231)得到的pet26b-marc种子液加入到含1l的lb液体培养基的试验容器中,在摇床内以37℃、200rpm震荡培养至含marc表达载体的浓度od600 = 0.9-1.1为止,随后将试验容器取出并置于冰浴中30 min,其中:
42、lb液体培养基中含卡那霉素,其浓度为50 ug/ml;
43、(233)向经过步骤(232)处理的菌液中分别加入硫酸亚铁铵、5-氨基乙酰丙酸、iptg,使得硫酸亚铁铵在菌液中的浓度为40 um,5-氨基乙酰丙酸在菌液中的浓度为0.25mm,iptg在菌液中的浓度为0.1 mm,然后混匀后放入摇床内以18℃、200rpm诱导培养16 h;
44、(234)将经过步骤(234)诱导完毕的菌液倒入离心杯,4000g离心15 min,弃上清液,沉淀表面用ddh2o清洗,随后加入30 ml上样缓冲液将沉淀菌体重悬至无明显菌块得到菌体重悬液,其中:
45、所述上样缓冲液的ph值为8.0;
46、所述上样缓冲液包括:100 mm tris-hcl、300 mm nacl、15 mm咪唑、10%v/v 甘油,余量为水;
47、(235)利用超生破碎仪将菌体重悬液破碎,其中:
48、变幅杆直径为6 mm;
49、破碎程序为:能量40%,超声循环工作时间2 s、暂停时间4 s,工作总时间30 min;
50、(236)将破碎后的菌液以10000g低温离心15min,取上清液即得到marc蛋白粗制裂解液;
51、步骤三:酶促反应体系的搭建
52、(31)向反应容器中分别加入下列物质:
53、,
54、其中:
55、所述nah2po4-na2hpo4缓冲液的浓度为1m,ph值为8.0;
56、所述(nh4)2so4储备液的浓度为2m;
57、(32)混匀后,震荡反应6-8h,震荡反应全程保持反应容器为敞口状态(即反应容器的上部氧气浓度与空气中的氧气浓度相同);
58、(33)分别向反应容器中加入600 ml步骤(23)制备的marc蛋白粗制裂解液、2.1gα-酮戊二酸、1.32 g抗坏血酸、适量的硫酸亚铁,使得硫酸亚铁在菌液中的浓度为0.1 mm,混匀后,继续震荡反应2h得到反应液;
59、步骤四:产物的分离与纯化
60、将步骤三得到的反应液用1n稀盐酸调节至ph=1,然后向其中加入1 l乙酸乙酯萃取至少三次,乳化层使用硅藻土过滤后分液,合并有机相并使用无水硫酸钠干燥,减压浓缩得到粗产物,然后将粗产物使用硅胶柱色谱进行纯化,dcm/meoh = 20:1条件下洗脱得到橙红色产物即得到细胞分裂抑制剂。
61、有益效果:本发明具有以下有益效果:
62、1.反应步骤少——在合成生物学研究成果基础上,开发了以关键酶laad、viob和marc为催化条件的2步合成方法;
63、2.以廉价且生物亲和性良好的l-色氨酸和l-组氨酸为底物,规避了现有方法中毒性化学品作为底物的参与,提高原子利用率、减小化学污染和可能引入的毒副作用;
64、3.在尽量小的反应体系内,实现克级产物的合成,并拥有高于目前化学合成手段的回收产率,节约后续此类药物制造的成本,降低药物价格。
- 还没有人留言评论。精彩留言会获得点赞!