吲哚吡唑类化合物及其制备方法和应用
本发明属于药物化学领域,具体涉及吲哚吡唑类化合物及其制备方法,并将其应用于以组蛋白赖氨酸特异性去甲基化酶(lsds,lsd1/lsd2)为靶点的创新药物研发。
背景技术:
1、组蛋白赖氨酸去甲基化酶(lsds)分为组蛋白赖氨酸特异性去甲基化酶1(lsd1)和组蛋白赖氨酸特异性去甲基化酶2(lsd2),是一类黄素腺嘌呤二核苷酸依赖的氧化酶,具有fad依赖的胺氧化酶结构域,能够特异性去除赖氨酸单双甲基化(journal of medicinalchemistry,2023,66,71–94)。研究表明;lsds在多种肿瘤细胞中过表达,与肿瘤的发生与发展密切相关(biomolecules,2022,12,462)。此外,lsds也与其他疾病如病毒感染、中枢神经系统疾病、心脑血管疾病等发生发展亦密切相关(epigenomics,2016,8,1103–1116;journal of hematology&oncology,2019,12,129)。
2、目前已有较多的lsds抑制剂报道,已报道的lsd1抑制剂较多,但尚无用于肿瘤治疗的上市lsd1抑制剂(journal of medicinal chemistry,2023,66,71–94)。目前处于临床研究阶段的lsd1抑制剂共有11个,lsd1抑制剂根据作用方式分为共价抑制剂和可逆抑制剂,目前处于临床研究阶段的主要是苯环丙胺类共价抑制剂,但该类lsd1共价抑制剂与fad不可逆共价结合,且对其他以fad为辅因子的多种蛋白也具有较好的亲和力,长期使用会产生毒性。可逆抑制剂是通过非共价的方式与fad结合,在安全性方面具有潜在的优势,其中cc-90011和sp-2577已进入i/ii期临床用于肿瘤治疗。相比之下,lsd2抑制剂报道较少,仍处于早期阶段,且已报道的抑制剂存在抑制活性低、选择性差等问题(breast cancer restreat,2014,146,99-108)。
技术实现思路
1、因此,本发明的一个目的在于提供一类吲哚吡唑类化合物及其制备方法,从而为寻找新型lsd1和lsd2抑制剂提供一条新途径。
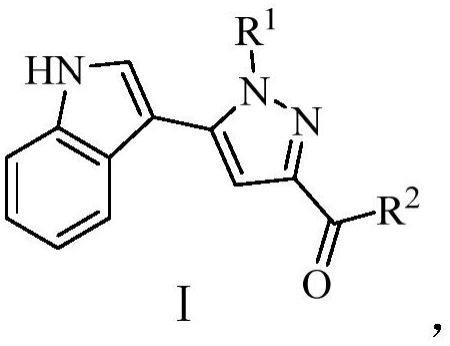
2、为实现上述目的,本发明提供一种吲哚吡唑类化合物,其包括吲哚吡唑类衍生物及其药学可接受的盐,上述吲哚吡唑类衍生物的通式如下所示:
3、
4、其中,上述通式i中的基团r1为氢原子、甲基、醛基、环己基、苄基、苯基、取代苯基或杂芳基;
5、所述通式中的基团r2为氮杂六元脂肪环、氮杂七元脂肪环、苯胺、取代苯胺基、n-取代c1~10链状胺基。
6、其中,本文中的“取代苯基”是指苯基上含有一个或多个取代基;优选地,该取代基可以为氰基、甲氧基、三氟甲基等。本文中的“杂芳基”是指苯环中至少一个c原子,且被n、s或o取代的芳香环基;优选为5-卤素吡啶。本文中的“取代苯胺基”是指苯胺基中的苯环上含有一个或多个取代基;优选地,该取代基可以为羟基、氨基、取代甲酰胺基、取代胺基、氮杂脂肪环基、苄胺基等;更优选地,该取代基可以为4-羟基、4-氨基、2-二甲胺基、4-二甲胺基、4-n,n-二甲酰胺基、哌嗪、n-甲基哌嗪基、n-苯基哌嗪基、4-甲基哌啶基、4-羟基哌啶基、4-甲基-4-氨基哌啶基、4-氨基哌啶基、吗啉基、n-哌啶基胺基、n-四氢吡喃基基胺基、四氢吡咯、四氢异喹啉基、4-(2-呋喃基亚甲基)-胺基。本文中的“n-取代c1~10链状胺基”优选为n-多取代c1~10胺基,更优选为n,n,n-三甲基乙二胺基、n,n-二甲基乙二胺基。
7、优选地,所述基团r1为h、甲基、醛基、苯基、4-三氟甲基苯基、4-腈基苯基、4-甲氧基苯基、苄基、环己基、5-溴吡啶基。
8、优选地,所述基团r2为吗啉基、哌嗪基、n,n-二甲基对苯二胺基、n-甲基哌嗪、4-氨基哌啶基、4-(4-甲基哌嗪)苯胺基、n,n-二甲基乙二胺基、苯氨基、4-氨基苯酚基、对苯二氨基、n-甲基高哌嗪基、4-二甲氨基哌啶基、4-羟基哌啶基、4-(4-吗啉基)苯胺基、n,n-二甲基乙二胺基、4-(1-吡咯烷基)苯胺基、n,n,n'-三甲基乙二胺基、4-(4-甲基-哌啶-1-基)-苯基胺基、n,n-二甲基邻苯二胺基、4-氨基-n,n-二甲基苯甲酰胺基、4-哌嗪基苯胺基、4-(4-甲基-哌啶-1-基)-苯基胺基、n-苯基哌啶-4-氨基、1-n-(氮杂环己烷-4-基)苯-1,4-二胺基、1-(4-氨基苯基)-4-羟基哌啶基。
9、上述吲哚吡唑类衍生物药学可接受的盐为三氟乙酸盐、硫酸盐、盐酸盐、甲磺酸盐等。
10、所述吲哚吡唑类衍生物为所述通式ⅰ所示的,且具有下列基团的化合物中的一个:
11、
12、
13、本发明提供一种吲哚吡唑类化合物的制备方法,包括以下步骤:
14、中间体c的制备:中间体b和肼试剂r1nhnh2发生反应,合成中间体c;其中,中间体b的结构通式为:中间体c结构通式为:
15、中间体d的制备:所述中间体c在碱性条件下发生水解反应,合成中间体化合物d,且该中间体化合物d的结构通式为
16、通式i的制备:中间体d和胺类化合物r2nh2在冰醋酸条件下发生缩合反应,合成通式i所示的吲哚吡唑类化合物。
17、所述中间体c的制备步骤包括:所述中间体b溶于冰醋酸中,然后缓慢滴加肼试剂r1nhnh2,加热回流合成中间体c,获得中间体c混合液,将中间体c混合液滴加至冰水中,伴随大量白色固体生成,经抽滤、干燥处理,得到所述中间体c。优选地,中间体b与肼类试剂r1nhnh2的摩尔比为1:2~1:3,加热回流搅拌1.5~3h。
18、所述中间体d的制备包括:先将所述中间体c溶于水中,再加入氢氧化钠,然后0~100℃下反应4~6h,其中,所述中间体c与氢氧化钠的摩尔比为1:0.5~1:5;采用tlc监测反应结束后,冷却至室温,用浓盐酸调节ph值1~2,经抽滤、干燥处理得到所述中间体d。
19、所述通式i的制备步骤包括:所述中间体d、胺类化合物r2nh2、缩合剂1-丙基磷酸酐(t3p)和有机碱dipea(中文名称:n,n-二异丙基乙胺)溶于n,n-二甲基甲酰胺中,常温下发生反应合成所述通式ⅰ所示的吲哚吡唑类化合物,获得通式i混合物;对所述通式i混合物进行提纯处理,获得通式i所示的吲哚吡唑类化合物。其中,优选地,所述中间体d、r2nh2、t3p和dipea摩尔比为1:0.5:0.5:0.5~1:3:3:3;反应时间6~8h。对所述通式i混合物进行提纯处理的方法包括:经tlc监测反应结束后,向所述通式i混合物中加入等量的水和乙酸乙酯进行萃取,有机相用水萃取三次,所述有机层经无水硫酸镁干燥后,抽滤,经柱层析分离,得到所述通式ⅰ所示的吲哚吡唑类化合物。
20、通式i所示的吲哚吡唑类化合物与药学可接受的酸发生反应,合成吲哚吡唑类化合物的药学可接受的盐,其中,所述药学可接受的酸为三氟乙酸、硫酸、盐酸或甲磺酸等。具体地,将所述通式ⅰ所示的吲哚吡唑类化合物溶于有机溶剂中,加入所述药学可接受的酸反应1~2h,如,三氟乙酸tfa,再除去其中的有机溶剂,制得所述吲哚吡唑类化合物的药学可接受的盐。
21、上述吲哚吡唑类化合物的制备方还包括中间体b的制步骤:将3-乙酰吲哚(化合物a)和草酸二乙酯在乙醇钠溶液中,于0~100℃反应,合成所述中间体b。具体地,将3-乙酰吲哚和草酸二乙酯按照摩尔比为1:1~1:2在乙醇钠溶液中于0~100℃搅拌2~3h,经tlc检测反应完全后,用浓盐酸调节ph至ph3~4,抽滤析出的固体,即可得到中间体b。如此,以3-乙酰吲哚为原料,合成目标化合物的路线如下所示:
22、
23、本发明还提供一种上述吲哚吡唑类化合物在制备lsds抑制剂中的应用。其中,lsds抑制剂为lsd1抑制剂、lsd2抑制剂或两者通用抑制剂。
24、因此,本案提供的上述吲哚吡唑类化合物是一类以吲哚和吡唑为基本骨架的化合物,经验证具有同时抑制lsd1和lsd2的作用,从而使得本发明为寻找一类新的基于lsds的创新药物发现开辟一条新途径。
- 还没有人留言评论。精彩留言会获得点赞!