一种砜吡草唑的制备方法与流程
本发明涉及砜吡草唑制备领域,尤其是涉及一种砜吡草唑的制备方法。
背景技术:
1、杂草严重影响小麦、高粱、玉米等农作物的丰产丰收,而使用除草剂是农田杂草防治中最经济、有效的手段。砜吡草唑是一种新型、广谱、高活性的苗前土壤处理除草剂,是由日本组合化学株式会社开发的优秀除草剂成分,具有应用广泛、生物活性高、施用量低、杀草谱广等优点。
2、砜吡草唑,英文名称为pyroxasulfone,cas号为447399-55-5,化学名称为[3-[(5-二氟甲氧基-1-甲基-3-三氟甲基吡唑-4-基)-甲基磺酰基]-4,5-二氢-5,5-二甲基-1,2-噁唑],分子式为c12h14f5n3o4s,相对分子质量:391.32。砜吡草唑为白色固体,熔点为157.6℃,沸点为362.4℃。其除草作用机理为,砜吡草唑属细胞分裂抑制剂,可抑制植物体内极长侧链脂肪酸vlcfa(c20-c30)的生物合成,进而阻碍分生组织和胚芽鞘的生长。砜吡草唑施用后,被杂草幼根与幼芽吸收,抑制幼苗早期生长,破坏分生组织与胚芽鞘,主要应用于玉米田、大豆田及小麦田等禾本科和阔叶杂草的防治。同时,有研究表明,砜吡草唑对麦田禾本科杂草鹅观草、多花黑麦草、雀麦、棒头草、蜡烛草,阔叶杂草大巢菜、播娘蒿、麦家公、泽漆等,均具有良好的防除效果,株抑制率和鲜重抑制率均可达到90%以上,应用前景广阔。
3、现有砜吡草唑的制备技术中,针对于重要中间体iii,通常需要经过环合、氯化、取代反应制得。但是在环合、氯化过程中,反应产物的收率指标不理想,且易于生成副产物,导致各环节需要多次洗涤、萃取及蒸馏脱溶,废液产生量高,能耗较大。并且,在氯化过程中,一般采用氯气、五氯化磷、三氯氧磷等氯化试剂,但氯气氧化性强,易腐蚀设备,生产危险性高;而五氯化磷、三氯氧磷在氯化过程中会产生大量含有磷酸盐的废液,废液处理难度大,处理成本高,环境污染问题突出。进一步的,前述缺陷还会影响最终制得的砜吡草唑的收率,增加砜吡草唑提纯难度,限制砜吡草唑纯度的进一步提升。
技术实现思路
1、为解决现有技术中存在的技术问题,本发明提供一种砜吡草唑的制备方法,能够有效克服环合、氯化过程中,反应产物的收率指标不理想,且易于生成副产物,导致各环节需要多次洗涤、萃取及蒸馏脱溶,以及生产危险性高,废液产生量大,后处理难度大的问题;有效降低生产过程中废液的产生量及处理难度,降低环境污染,同时,进一步提升制得的砜吡草唑的收率及纯度。
2、为解决以上技术问题,本发明采取的技术方案如下:
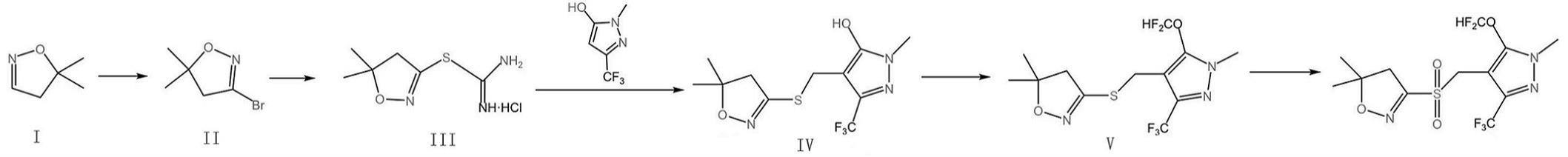
3、一种砜吡草唑的制备方法,所述制备方法由以下步骤组成:制备化合物ii、制备化合物iii、制备化合物iv、制备化合物v、制备砜吡草唑。
4、所述制备化合物ii,将化合物i5,5-二甲基-4,5-二氢异噁唑138.8-178.4g(1.4-1.8mol)投入至正己烷中700-900ml中,分散均匀后,降温至3-5℃,保温;在120-200rpm搅拌条件下,以0.2-0.5ml/min的滴加速率,滴入浓度为60-65wt%的氢溴酸90-95g,滴加完成后,继续搅拌20-50min;氢溴酸滴加及搅拌过程中,保持温度为3-5℃;投入催化剂,30-60rpm搅拌条件下,以0.8-1ml/min的滴加速率,滴入氯化溴178.7-224.8g(1.55-1.95mol),滴加完成后,保温搅拌3-4h后,滤除固体物,静置分层;采用1-1.2倍体积的正己烷提取酸层后,合并有机层,采用2-3倍体积的去离子水洗涤有机层,制得含有化合物ii的正己烷溶液。
5、所述制备化合物ii中,化合物i的结构式为:
6、;
7、化合物ii的结构式为:
8、。
9、所述制备化合物ii中,催化剂的添加量为化合物i重量的5-7wt%;
10、催化剂由以下步骤制得:活化、吸附。
11、所述活化,将活性炭投入至8-10倍体积的磷酸中,升温至40-50℃,保温搅拌3-5h,滤出并采用8-10倍体积的去离子水洗涤2-3次后;将活性炭转入至高压反应釜内,通入二氧化碳,升压至10-12mpa,保持压力并升温至70-80℃,保温保压2-3h后,自然冷却,泄压;将活性炭转入恒温箱内,70-80℃静置2-4h,制得活化活性炭。
12、所述活化中,磷酸浓度为1-1.5mol/l;
13、活性炭的粒径为2-3mm,孔径为8-15nm。
14、所述吸附,将活化活性炭投入至8-10倍体积的吸附液中,氮气气氛环境下,升温至30-40℃,保温回流搅拌2-4h后,滤出固体物;采用5-8倍体积的无水己烷洗涤固体物3-4次,真空干燥去除无水己烷,制得催化剂。
15、所述吸附中,吸附液为溶解有三乙基铝、二氯化乙基铝的甲苯溶液;吸附液中,三乙基铝浓度为2-3wt%,二氯化乙基铝浓度为4.5-6.0wt%。
16、所述制备化合物iii,向所述含化合物ii的正己烷溶液中,加入浓度为32-36wt%的盐酸42-54g,搅拌20-30min后;保持搅拌并投入硫脲110.4-140.8g,控制温度为22-28℃,搅拌4-6h;降温至2-6℃,保温结晶2-3h后,滤出结晶物,采用0.8-1倍体积的正己烷淋洗结晶物后,50-55℃干燥8-10h,制得化合物iii。
17、所述化合物iii的结构式为:
18、。
19、所述制备化合物iv,将三氟乙酰乙酸乙酯184-202.4g投入至1.8-2.2倍重量份的乙酸中,分散均匀,降温至0-2℃,保温滴加浓度为40wt%的甲基肼水溶液128-140.8g,甲基肼水溶液滴加过程中,保持温度为0-5℃;滴加完成后,以0.1-0.15℃/min的升温速率,升温至75-80℃,保温回流搅拌6-8h;继续加入600-700ml去离子水,搅拌40-60min,滤出固体物,采用6-8倍体积的去离子水洗涤3-4次,制得中间体1-甲基-3-(三氟甲基)-1h-吡唑-5-醇;然后将中间体1-甲基-3-(三氟甲基)-1h-吡唑-5-醇79.7-84.7g与氢氧化钠68-72g投入至450-550ml的水中,搅拌溶解后,以1-2g/min的滴加速率,滴入浓度为32wt%的甲醛溶液104-115g,滴加完成后,继续搅拌30-40min,升温至28-30℃,保温搅拌并以2-3ml/min的滴加速率,滴入化合物iii的水溶液303.8-322.8g,保温回流搅拌7-8h后,采用盐酸调节ph值至6-6.5,静置至无固体析出,滤出固体物,采用8-10倍体积的去离子水洗涤固体物,真空干燥至恒重,制得化合物iv。
20、所述制备化合物iv中,化合物iii水溶液的浓度为25wt%。
21、化合物iv的结构式为:
22、。
23、所述制备化合物v,室温条件下,将化合物iv100-108.9g投入至5-6倍重量份室温n,n-二甲基甲酰胺中,搅拌15-30min后,继续投入130-135g碳酸钾,搅拌40-80min后;在搅拌条件下,以2-2.5g/min的投入速率,通入二氟一氯甲烷138.4-147.1g,通入完成后继续搅拌7-8h;继续加入1.5-2倍dmf重量份的去离子水,以及0.5-0.6倍dmf重量份的1,1-二氯甲烷,搅拌30-60min后,依次采用去离子水、饱和氯化钠溶液洗涤,分离出有机相,真空蒸馏脱溶,制得化合物v。
24、所述化合物v的结构式为:
25、。
26、所述制备砜吡草唑,室温条件下,将化合物v52.6-60.1g投入至2.5-3.5倍重量份的甲醇中,分散均匀后,继续投入二水合钨酸钠2.2-2.5g和浓度为98%的浓硫酸1-1.2ml;搅拌10-20min后,以1-2g/min的滴加速率,滴加浓度为31%的双氧水52-56g,滴加完成后,继续搅拌5-6h后,加入0.3-0.4倍重量份的去离子水,冷却至2-5℃,静置至无固体物析出,滤出固体物,并采用同等温度的1.2-1.5倍重量份的甲醇淋洗2-3次,真空干燥,制得砜吡草唑。
27、与现有技术相比,本发明的有益效果为:
28、(1)本发明的砜吡草唑的制备方法,通过在制备化合物ii步骤中,采用氯化溴替代常规氯化试剂;同时配合经活化、吸附步骤制得的催化剂,通过将活化处理后的活化活性炭对吸附液中有效成分进行吸附,实现对化合物ii的有效催化;各技术手段相互配合,有效克服传统砜吡草唑的环合、氯化过程中,反应产物的收率指标不理想,易于生成副产物,各环节需要多次洗涤、萃取及蒸馏脱溶,以及生产危险性高,废液产生量大,后处理难度大的问题;有效降低生产过程中废液的产生量及处理难度,降低环境污染;同时,提升中间各化合物的纯度及收率指标,进一步提升制得的砜吡草唑的收率及纯度。
29、(2)本发明的砜吡草唑的制备方法,制备化合物ii步骤中,制得的化合物ii的收率为99.3-99.7%;制备化合物iii步骤中,制得的化合物iii纯度为98.0-98.4wt%,收率为93.9-95.0%;有效保证后续化合物iv-v、砜吡草唑纯度及收率的进一步提升。
30、(3)本发明的砜吡草唑的制备方法,有效抑制副产物的产生,简化提纯精制环节,降低配套装置及生产能耗,减少生产废液的生产量;同时,有效避免采用氯气作为氯化剂可能的设备腐蚀、生产危险性,提高生产装置使用寿命及维保频率;无需采用五氯化磷、三氯氧磷,避免氯化过程中产生的大量含有磷酸盐废液,消除相关废液处理成本,降低环境污染;本发明的砜吡草唑的制备方法生产安全性好,反应废液易于处理,有效降低生产设备及环保成本。
31、(4)本发明的砜吡草唑的制备方法,制得的砜吡草唑纯度可达99.5-99.6wt%,综合收率可达62.0-66.0%。
- 还没有人留言评论。精彩留言会获得点赞!