一种基于TadA8e突变体实现碱基颠换的编辑工具及应用的制作方法
本发明涉及基因编辑,具体涉及一种实现水稻g-c碱基颠换的编辑工具ccdc-cgbe及应用
背景技术:
1、碱基编辑是在不产生dna双链断裂的情况下实现单核苷酸靶向突变的技术。目前已开发的碱基编辑系统主要包括胞嘧啶碱基编辑器(cbes)、腺嘌呤碱基编辑器(abes)和鸟嘌呤碱基编辑器(cgbes)等。abe和cbe系统主要产生嘧啶到嘧啶或嘌呤到嘌呤碱基的转变,无法实现嘧啶与嘌呤如c-g、a-t之间的碱基颠换。研究发现,cbe系统除诱导c-t碱基转换外,特定条件下,如剔除融合蛋白中的尿嘧啶糖基化酶抑制剂(ugi)组分会诱导部分位点发生c-a和c-g碱基颠换,在该思路的指引之下cgbe碱基编辑器得以开发。2020年,张学礼和毕昌昊实验室以及j.keith joung和julian grünewald实验室在相同思路的指引下,通过改造现有的单碱基编辑工具cbe,构建出哺乳动物细胞适用的介导c-g碱基颠换的新工具gbe/cgbe。cgbe碱基编辑器主要包括cas9切口酶(ncas9-d10a)、胞苷脱氨酶和尿嘧啶-n-糖基化酶(ung)三部分。这一系列新工具的出现让单碱基编辑再添新武器,也为后续的基础研究和临床转化提供了新的助力。
2、然而现有的cbes/cgbes均依赖于天然的胞嘧啶脱氨酶aid/apobec家族成员,由于胞嘧啶脱氨后会激活碱基切除修复通路,不可避免的会产生较高的indels副产物。主要表现在两个方面:cbe除了将c转换为t以外,还有一定的几率将c转变为g/a,这是由于udg可以将碱基u切除形成无嘧啶无嘌呤的位点ap,在ap裂解酶或自发断裂下产生一个缺口,进而与ncas9在非编辑链产生的缺口刚好形成一个dsb,经过nhej修复途径产生indels.更为重要的是现有的cbe会产生显著的不依赖于cas9的dna和rna随机脱靶,而且较宽的编辑窗口引起难以避免的旁观者效应,在安全性等方面存在隐患。与cbe/cgbes不同,abe使用从tada(大肠杆菌中的一种转移rna(trna)腺嘌呤脱氨酶)进化而来的非天然腺嘌呤脱氨酶,以非常高的产品纯度(超过99.9%)、最少的indels和相对紧凑的编辑窗口诱导dna中a-g的转换。2019年,sangsu bae/jin-soo kim研究团队在nbt上发表的研究论文表明,腺嘌呤碱基编辑还在狭窄的编辑窗口(c5-c7)和受限的tc*n中将胞嘧啶转化为鸟嘌呤或胸腺嘧啶;其中,腺嘌呤碱基编辑诱导的胞嘧啶效率高达11.2%,这表明进化的tada具有胞嘧啶脱氨能力;而且不同版本abe碱基编辑器转化效率是有差异的,表明各种tada突变对碱基编辑特异性的影响不同。基于此,2022年华东师范大学李大力和哈佛大学broad研究所david r.liu实验室分别使用不同的方法对腺嘌呤脱氨酶tada-8e重新设计改造,开发了系列消除固有的腺嘌呤脱氨酶活性的新型td-cbes(tada-derived cytosine base editor),展现出非常低的indels事件,创新性地构建了一系列不依赖天然胞嘧啶脱氨酶家族的“精准且安全”的新型胞嘧啶碱基编辑器。
3、在植物中,产生更多的碱基替代类型可以扩大其应用,并有助于创造新的种质资源。在之前的研究中,基于第一代cgbes,yiping qi及陆钰明/朱健康研究组在水稻、番茄和白杨物种中实现了碱基c-g的颠换,这种cgbes为植物的c-to-g碱基编辑带来了巨大的希望,但是效率较低并且伴随大量的副产物难以在植物中推广应用,因此本发明希望在植物中建立基于腺嘌呤脱氨酶tada变体的高效cgbe碱基编辑器,在实现更高编辑效率的同时,保持基因编辑的精准性,降低副产物的产生,为植物育种以及创制新的种质资源提供有力工具。
技术实现思路
0、
技术实现要素:
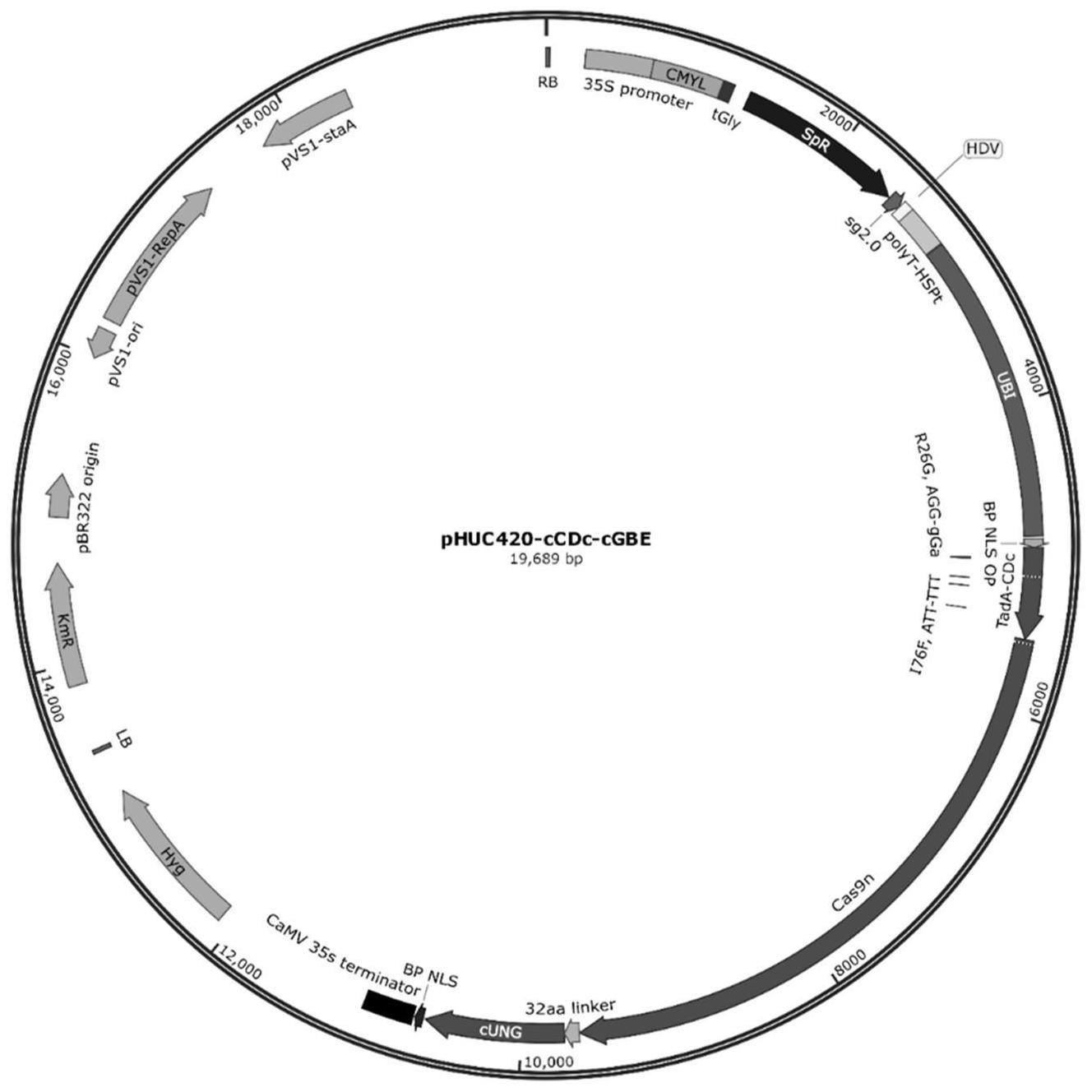
1、针对上述问题,本发明提供了一种基于tada8e突变体(r26g/e27r/v28g/i76f/h96n/m151i,将该变体命名为tada-cdc),融合鳕鱼源的尿嘧啶dna糖基化酶cung,构建新型的cgbe碱基编辑器ccdc-cgbe,实现由g-c的高效碱基颠换。
2、本技术的发明人在水稻基因研究过程中发现了一种tada8e突变体,发明人发现,将该突变体与融合鳕鱼源的尿嘧啶dna糖基化酶cung配合使用,构建碱基编辑工具,可以高效地在水稻中实现碱基颠换。
3、具体而言,本发明提供一种基于tada8e突变体实现水稻g-c碱基编辑器。实验中,发明人针对tada8e进行6个特定氨基酸的点突变,并且进行水稻密码子优化,融合水稻密码子优化的失活型spcas9(nspcas9)以及鳕鱼源的尿嘧啶dna糖基化酶cung,得到tada-cdc-nspcas9-cung表达框,将该表达框整合到植物表达载体中构建相应的打靶载体,而后通过水稻遗传转化实现对水稻特异基因编辑。
4、在第二个方面,本发明提供一种含有所述tada-cdc-nspcas9-cung表达框的植物表达载体。该植物表达载体的构建方法是利用psti/saci酶切位点,用psti/saci酶切phucspr载体并回收,由于tada-cdc-nspcas9-cung基因两端加有psti/saci酶切位点,可以利用t4连接酶将tada-cdc-nspcas9-cung连接到phuc spr载体,得到植物中间表达载体phuc-tada-cdc-nspcas9-cung;进一步地,通过hindiii酶切位点引入35s cmylcv spr sg2.0polyt复合型启动子表达框,得到植物打靶载体phuc420-tada-cdc-nspcas9-cung,命名为ccdc-cgbe。
5、另一方面,本发明在表达载体的基础上,根据实验的实际需要,构建相应的基因打靶载体。
6、另一方面,本发明还提供一种表达盒,所述表达盒中包含上述的tada-cdc-nspcas9-cung基因。
7、另一方面,本发明还提供一种上述基因、表达盒或载体的应用,所述应用包括利用所述tada-cdc-nspcas9-cung基因完成水稻体内g-c的碱基颠换,获得带有突变位点的转基因植物或植物部分。
8、在另一个方面,本发明提供一种利用ccdc-cgbe表达载体将打靶载体导入水稻细胞的方法,利用ccdc-cgbe表达载体在表达载体的基础上只需进行简单的退火、酶切连接作用即可获得特异基因的打靶载体(ccdc-cgbe-acc),然后将打靶载体导入水稻细胞。
9、该方法包括下述步骤:
10、(1)将日本晴水稻种子去壳、灭菌后将胚分离出来,置于愈伤组织诱导培养基上以产生次级愈伤组织;
11、(2)将次级愈伤组织转移至新的愈伤组织诱导培养基预培养;
12、(3)将步骤(2)中获得的愈伤组织与携带ccdc-cgbe-acc的打靶载体的农杆菌充分接触15分钟;
13、(4)将步骤(3)的愈伤组织转移到其上垫有三张无菌滤纸(加入2.5-3.5ml农杆菌悬浮培养基)的培养皿中,21-23℃培养48小时;
14、(5)将步骤(4)的愈伤组织置于前筛选培养基上培养5-7天;
15、(6)将步骤(5)的愈伤组织转移筛选培养基上,以获得抗性愈伤组织;
16、(7)将抗性愈伤组织转移到分化再生培养基中分化成苗;和
17、(8)将步骤(7)的苗转移到生根培养基中生根。
18、其中所述步骤(1)中的种子是日本晴成熟种子;所述步骤(1)、(2)中的诱导培养基是说明书表1所列出的诱导培养基;所述步骤(3)中的与农杆菌接触是将愈伤组织浸泡在所述农杆菌悬浮液中;所述步骤(4)中的农杆菌悬浮培养基是说明书表1所列出的悬浮培养基;所述步骤(5)中的前筛选培养基是说明书表1所列出的前筛选培养基;所述步骤(6)中的筛选培养基是说明书表1所列出的筛选培养基;所述步骤(7)中的分化再生培养基是说明书表1所列出的分化再生培养基;所述步骤(8)中的生根培养基是说明书表1所列出的生根培养基。
19、另一方面,本发明提供一种利用所述的编辑工具获得编辑植株的方法,所述方法包括:
20、(1)将日本晴水稻种子去壳、灭菌后将胚分离出来,置于愈伤组织诱导培养基上以产生次级愈伤组织;
21、(2)将次级愈伤组织转移至新的愈伤组织诱导培养基预培养;
22、(3)将步骤(2)中获得的愈伤组织与携带ccdc-cgbe-acc的打靶载体的农杆菌充分接触15分钟;
23、(4)将步骤(3)的愈伤组织转移到2.5-3.5ml农杆菌悬浮培养基上,21-23℃培养;
24、(5)将步骤(4)的愈伤组织置于前筛选培养基上培养5-7天;
25、(6)将步骤(5)的愈伤组织转移筛选培养基上,以获得抗性愈伤组织;
26、(7)将抗性愈伤组织转移到分化再生培养基中分化成苗;和
27、(8)将步骤(7)的苗转移到生根培养基中生根,
28、优选地,上述步骤中,以loc_os05g22940中的核苷酸序列
29、cctcgctaactggagaggcttct为打靶位点。
30、在优选的实施方案中,其中所述水稻是粳稻,更优选地,所述水稻是粳稻日本晴。
31、
32、表1培养基的示例性配方
33、表格中所提到的“优化的n6大量元素”指的是,该n6大量元素中[no3-]/[nh4+]=40mm/10mm。
34、在优选的实施方案中,所述tada-cdc-nspcas9-cung的核苷酸序列为seq id no:1所示的核苷酸序列,具体如下:
35、
36、
37、
38、
- 还没有人留言评论。精彩留言会获得点赞!