一种甘氨酸-吡咯烷类化合物及其制备方法和应用
本发明属于药物合成,涉及一种吡咯烷类化合物,特别是涉及一种甘氨酸-吡咯烷类化合物,该类化合物的制备方法以及该类化合物在药物中的应用。
背景技术:
1、成纤维细胞活化蛋白(fap)是一种ii型跨膜丝氨酸蛋白水解酶,存在于肿瘤微环境的激活成纤维细胞中,在恶性肿瘤的迁移、增殖、侵袭、化疗耐药性和免疫抑制等方面发挥重要作用。fap在卵巢癌、结肠癌、乳腺癌等多种肿瘤及关节炎症中差异性高表达,在正常组织的静止成纤维细胞中不表达。fap的这种特异性分布,使其有望成为肿瘤临床诊断和临床治疗的一个重要靶标,具有极高的研究价值,发展fap特异性抑制剂已经是肿瘤靶向药物开发的热点。
2、近年来,以fap作为靶点的研究主要集中在fap抑制剂与抗fap单克隆抗体上,但其临床效果并不理想,药物治疗成本也及其高昂,从而使其的应用受限。
3、目前针对fap的抑制剂研究还处于初始阶段,抑制fap的天然产物亦少有报道,多数为合成的fap抑制剂,临床上还没有靶向fap治疗肿瘤的上市药物。
4、talabostat是第一个进入临床实验的fap小分子抑制剂。然而,除了对fap显示出明显的抑制作用外,talabostat对二肽基肽酶的其他成员也显示出一定的抑制能力,表明其虽然是一种良好的靶向fap肿瘤治疗药物,但在fap活性选择性上还存在一定缺陷,有待进一步研究。同时,talabostat也有一些副作用,其中大部分与细胞因子释放相关,最常见的不良反应是水肿。
5、为了改善talabostat的不足,poplawski s e等(identification of selectiveand potent inhibitors of fibroblast activation protein and prolyloligopeptidase [j]. journal of medicinal chemistry, 2013, 56(9): 3467-3477.)报道了多种基于下述结构的化合物compound 4作为先导化合物的fap抑制剂。
6、
7、然而,这些fap抑制剂虽然对compound 4的吡咯氰基部分进行了延长,但并没有很好地占据fap蛋白的活性空腔,要么抑制活性不高,要么对fap的靶向性较差,或者没有解决药物的不良反应。因此,继续对此类化合物进行结构改造,开发一类对成纤维细胞活化蛋白具有良好抑制活性的抑制剂,具有重要的现实意义。
技术实现思路
1、本发明的目的是针对现有技术的不足,提供一种甘氨酸-吡咯烷类化合物及其制备方法和应用。
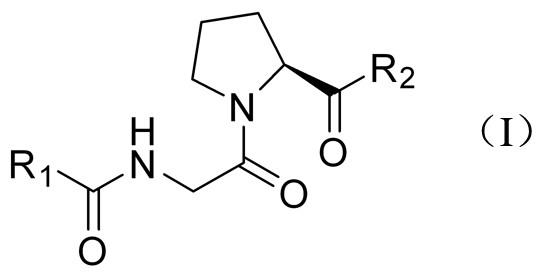
2、本发明首先提供了一种甘氨酸-吡咯烷类化合物,其具有以下结构通式(i)所示的结构:
3、
4、其中,r1为或;r2为、或。
5、更具体地,其中的r3选自氢、c1-4烷基、c1-4烷氧基、c1-4卤代烷基、硝基、三氟甲基中的任意一种。
6、更优选地,其中的r3选自氢、甲基、甲氧基、溴甲基、硝基、三氟甲基中的任意一种。
7、更具体地,其中的r4选自氢、羟基、卤素、c1-4卤代烷基中的任意一种。
8、更优选地,其中的r4选自氢、羟基、溴甲基中的任意一种。
9、更进一步地,本发明所述的甘氨酸-吡咯烷类化合物可以是以下具体结构式的化合物中的任意一种:
10、,简称i-a-1;
11、,简称i-a-2;
12、,简称i-a-3;
13、,简称i-a-4;
14、,简称i-a-5;
15、,简称i-a-6;
16、,简称i-b-1;
17、,简称i-b-2;
18、,简称i-b-3;
19、,简称i-b-4;
20、,简称i-b-5;
21、,简称i-b-6;
22、,简称i-c-1;
23、,简称i-c-2;
24、,简称i-c-3;
25、,简称i-c-4;
26、,简称i-c-5;
27、,简称i-c-6;
28、,简称i-c-7;
29、,简称i-d-1;
30、,简称i-d-2;
31、,简称i-d-3;
32、,简称i-d-4;
33、,简称i-d-5;
34、,简称i-d-6;
35、,简称i-e-1;
36、,简称i-e-2;
37、,简称i-e-3;
38、,简称i-e-4;
39、,简称i-e-5;
40、,简称i-f-1;
41、,简称i-f-2;
42、,简称i-f-3;
43、,简称i-f-4;
44、,简称i-f-5;
45、,简称i-f-6。
46、进而,本发明还给出了一种典型的所述甘氨酸-吡咯烷类化合物的制备方法。
47、需要说明的是,以下提供的甘氨酸-吡咯烷类化合物的制备方法并非是用于制备该化合物的唯一合成路线,还可以采用其他适合的合成路线来制备本发明所述的甘氨酸-吡咯烷类化合物,本发明对其没有特别的限定。
48、首先,是以下述式(ii)所示的酰氯化合物与甘氨酸乙酸乙酯在碱催化下进行酰胺反应,得到下述式(iii)所示的第一中间体:
49、,
50、;
51、其次,再以所述第一中间体在碱性条件下发生水解反应得到下述式(iv)所示的第二中间体:
52、;
53、同时,是以l-脯氨酸先在五氯化磷作用下生成酰氯,再在路易斯酸催化下与结构r2h所示的化合物发生付-克反应,得到下述式(v)所示的第三中间体:
54、;
55、最后,以所述第二中间体与第三中间体进行hatu缩合反应,制备得到式(i)所示的甘氨酸-吡咯烷类化合物。
56、本发明上述制备方法的合成路线可以以下述反应方程式表示:
57、
58、
59、
60、具体地,本发明所述制备方法中,所述酰胺反应是在二氯甲烷溶剂体系中,室温下以三乙胺作为催化剂进行反应的,反应时间优选为4-6h。
61、具体地,本发明所述制备方法中,所述水解反应是在甲醇与水的混合溶液体系中,加热至40-50℃进行的,反应时间优选为16-20h。
62、具体地,本发明所述制备方法中,所述l-脯氨酸与五氯化磷先在无水二氯甲烷中0-4℃下反应2-3h生成酰氯,除去溶剂后,残余物中加入路易斯酸和r2h,升温至60-64℃进行付-克反应,反应时间优选为4-6h。
63、具体地,本发明所述制备方法中,所述第二中间体与第三中间体是在溶剂dmf中dipea存在下,室温下以hatu催化进行缩合反应4-5h,制备式(i)所示的甘氨酸-吡咯烷类化合物。
64、其中,dipea化学名称n-乙基-n-异丙基丙烷-2-胺,hatu化学名称2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯。
65、进一步地,本发明中所述式(ii)所示的酰氯化合物可以是以结构为r1h的芳香酸与草酰氯反应得到。
66、更进一步地,基于本发明所述甘氨酸-吡咯烷类化合物对成纤维细胞活化蛋白具有的抑制作用,本发明还提供了所述化合物的医药用途。
67、具体地,本发明是提供了所述甘氨酸-吡咯烷类化合物在制备成纤维细胞活化蛋白抑制剂中的应用。
68、本发明所述的甘氨酸-吡咯烷类化合物是基于先导化合物compound 4为基础,针对现有甘氨酸-氰基吡咯烷类化合物fap抑制剂的结构进行进一步研究,在保留甘氨酸-吡咯烷结构的前提下,引入了带有不同取代基的芳香基团,这些芳香基团可以很好地占据fap蛋白的活性空腔,使得甘氨酸-吡咯烷能够与fap蛋白的活性位点更好地结合,并通过生物电子等排原理引入含刚性结构的羰基取代原有的氰基,增强了化合物的抑制活性功能。
69、本发明利用分子对接和3d-qsar技术,通过对先导化合物compound 4的结构进行进一步优化,设计、合成了36个含有羰基的甘氨酸-吡咯烷类fap抑制剂。新的系列抑制剂立体结构稳定,而且能稳定占据活性位点,具有称为新一代抗癌药物或作为新的先导化合物获得性能更优异的成纤维细胞活化蛋白抑制剂的潜力。
70、本发明提供的甘氨酸-吡咯烷类化合物制备方法简单,易于大批量制备,价格低廉。
71、实施方式
72、下面结合实施例对本发明的具体实施方式作进一步的详细描述。以下实施例仅用于更加清楚地说明本发明的技术方案,从而使本领域技术人员能很好地理解和利用本发明,而不是限制本发明的保护范围。
73、本发明实施例中涉及到的生产工艺、实验方法或检测方法,若无特别说明,均为现有技术中的常规方法,且其名称和/或简称均属于本领域内的常规名称,在相关用途领域内均非常清楚明确,本领域内技术人员能够根据该名称理解常规工艺步骤并应用相应设备,按照常规条件或制造商建议的条件进行实施。
74、本发明实施例中使用的各种仪器、设备、原料或试剂,并没有来源上的特殊限制,均为可以通过正规商业途径购买获得的常规产品,也可以按照本领域技术人员熟知的常规方法进行制备。
75、实施例
76、实施例1:以苯甲酸为原料合成抑制剂i-a-1
77、将苯甲酸(5.0mmol)加入到二氯甲烷(20ml)中,滴加无水dmf(0.5ml)和草酰氯(5.0ml),室温下搅拌20h制备产物ii。
78、以旋转蒸发仪除去溶剂和多余的草酰氯,剩余残渣用二氯甲烷(20ml)溶解,加入甘氨酸乙酸乙酯盐酸盐(306mg,5.0mmol)和三乙胺(2.5ml,15mmol),室温下搅拌4h,用稀盐酸溶液洗涤,形成产物iii沉淀。
79、然后将氢氧化钠(5.0mmol)加入含有产物iii的甲醇(15.0ml)与水(15.0ml)的混合溶液中,搅拌反应16h,以旋转蒸发仪除去溶剂,将残余物重溶于水中,稀盐酸洗涤酸化,析出固体产物,低温过滤、干燥,制备得到产物iv。
80、
81、将l-脯氨酸(0.667g,5.79mmol)和五氯化磷(1.206g,5.79mmol)加入到含有二氯甲烷(17.0ml)的25ml茄形瓶中,低温反应器中0℃下搅拌反应2h。用旋转蒸发仪将溶剂蒸发除去后,室温下干燥1h。
82、残渣中加入苯(17.0ml)和氯化铝(2.316g,17.37mmol),于60℃加热反应4h,冷却至室温,倒入1m hcl溶液与碎冰的混合物中,分离出水相,以过量碳酸钠中和,加入适量二氯甲烷,过滤,少量二氯甲烷洗涤碳酸钠滤饼,合并有机相,旋转蒸发仪旋干后,经柱色谱分离,得到产物v。
83、
84、将产物iv(53mg,0.3mmol)溶于3ml dmf中,加入dipea(0.610ml,1.5mmol)和hatu(73mg,0.3mmol),15min后加入到含有产物v(52.5mg,0.3mmol)的dmf溶液(0.5mmol)中,室温下搅拌反应3-4h。旋转蒸发仪旋蒸除去溶剂,残渣溶解在乙酸乙酯中,以饱和碳酸氢钠和盐酸溶液洗涤后,硫酸钠干燥,过滤、蒸发,以二氯甲烷-甲醇柱层析纯化,得到抑制剂i-a-1目标产物49mg,产率49%。
85、
86、1h nmr (600 mhz, cdcl3) δ 8.16 (d, j = 7.3 hz, 1h), 7.81 (dd, j = 7.1,4.5 hz, 2h), 7.59 (dd, j = 15.2, 7.6 hz, 2h), 7.49 (q, j = 6.3 hz, 3h), 7.39(t, j = 7.7 hz, 3h), 5.33 (d, j = 4.7 hz, 1h), 3.82 – 3.77 (m, 2h), 3.68 –3.60 (m, 2h), 2.19 (dd, j = 16.8, 9.0 hz, 2h), 2.10 (dt, j = 7.5, 4.2 hz,2h), 3.00 (dd, j = 16.8, 3.7 hz, 1h), 2.81 (dd, j = 16.8, 10.9 hz, 1h), 1.20(t, j = 7.1hz, 3h)。
87、13c nmr (151 mhz, cdcl3) δ 196.74, 170.83, 163.25, 141.83, 134.47,133.95, 133.38, 129.50, 128.90, 128.56, 126.84, 63.39, 61.15, 42.23, 31.45,19.01。
88、ms esi ( m/z): calcd for c20h21n2o3 [m+h]+: 337.15,found 337.03。
89、实施例2:以4-甲基苯磺酰氯为原料合成抑制剂i-b-1
90、将4-甲基苯磺酰氯(5.0mmol)和甘氨酸乙酸乙酯盐酸盐(306mg,5.0mmol)、三乙胺(2.5ml,15mmol)加入二氯甲烷(20ml)中,室温下搅拌4h,用稀盐酸溶液洗涤,形成产物iii沉淀。
91、然后将氢氧化钠(5.0mmol)加入含有产物iii的甲醇(15.0ml)与水(15.0ml)的混合溶液中,搅拌反应16h,以旋转蒸发仪除去溶剂,将残余物重溶于水中,稀盐酸洗涤酸化,析出固体产物,低温过滤、干燥,制备得到产物iv。
92、
93、将l-脯氨酸(0.667g,5.79mmol)和五氯化磷(1.206g,5.79mmol)加入到含有二氯甲烷(17.0ml)的25ml茄形瓶中,低温反应器中0℃下搅拌反应2h。用旋转蒸发仪将溶剂蒸发除去后,室温下干燥1h。
94、残渣中加入苯(17.0ml)和氯化铝(2.316g,17.37mmol),于60℃加热反应4h,冷却至室温,倒入1m hcl溶液与碎冰的混合物中,分离出水相,以过量碳酸钠中和,加入适量二氯甲烷,过滤,少量二氯甲烷洗涤碳酸钠滤饼,合并有机相,旋转蒸发仪旋干后,经柱色谱分离,得到产物v。
95、
96、将产物iv(58mg,0.3mmol)溶于3ml dmf中,加入dipea(0.610ml,1.5mmol)和hatu(73mg,0.3mmol),15min后加入到含有产物v(53mg,0.3mmol)的dmf溶液(0.5mmol)中,室温下搅拌反应3-4h。旋转蒸发仪旋蒸除去溶剂,残渣溶解在乙酸乙酯中,以饱和碳酸氢钠和盐酸溶液洗涤后,硫酸钠干燥,过滤、蒸发,以二氯甲烷-甲醇柱层析纯化,得到抑制剂i-b-1目标产物29mg,产率27%。
97、
98、1h nmr (600 mhz, cdcl3) δ 9.35 (s, 1h), 7.99 (t, j = 6.9 hz, 2h), 7.80(d, j = 8.1 hz, 1h), 7.71 – 7.68 (m, 2h), 7.52 – 7.44 (m, 2h), 7.18 (d, j =7.8 hz, 2h), 5.70 – 5.46 (m, 1h), 4.22 (s, 2h), 3.79 – 3.72 (m, 2h), 2.37 –2.34 (m, 3h), 2.14 – 2.09 (m, 2h), 2.04 – 1.95 (m, 2h)。
99、13c nmr (151 mhz, cdcl3) δ 196.74, 170.83, 163.25, 141.83, 134.47,133.95, 133.38, 129.50, 128.90, 128.56, 126.84, 63.39, 61.15, 42.23, 31.45,19.01, 13.65。
100、ms esi ( m/z): calcd for c21h21n2o3 [m-h]—: 349.38,found 349.16。
101、实施例3:以4-甲基苯磺酰氯为原料合成抑制剂i-b-5
102、将4-甲基苯磺酰氯(5.0mmol)和甘氨酸乙酸乙酯盐酸盐(306mg,5.0mmol)、三乙胺(2.5ml,15mmol)加入二氯甲烷(20ml)中,室温下搅拌4h,用稀盐酸溶液洗涤,形成产物iii沉淀。
103、然后将氢氧化钠(5.0mmol)加入含有产物iii的甲醇(15.0ml)与水(15.0ml)的混合溶液中,搅拌反应16h,以旋转蒸发仪除去溶剂,将残余物重溶于水中,稀盐酸洗涤酸化,析出固体产物,低温过滤、干燥,制备得到产物iv。
104、
105、将l-脯氨酸(0.667g,5.79mmol)和五氯化磷(1.206g,5.79mmol)加入到含有二氯甲烷(17.0ml)的25ml茄形瓶中,低温反应器中0℃下搅拌反应2h。用旋转蒸发仪将溶剂蒸发除去后,室温下干燥1h。
106、残渣中加入萘(0.717g)和氯化铝(2.316g,17.37mmol),于60℃加热反应4h,冷却至室温,倒入1m hcl溶液与碎冰的混合物中,分离出水相,以过量碳酸钠中和,加入适量二氯甲烷,过滤,少量二氯甲烷洗涤碳酸钠滤饼,合并有机相,旋转蒸发仪旋干后,经柱色谱分离,得到产物v。
107、
108、将产物iv(58mg,0.3mmol)溶于3ml dmf中,加入dipea(0.6ml,1.5mmol)和hatu(73mg,0.3mmol),15min后加入到含有产物v(68mg,0.3mmol)的dmf溶液(0.5mmol)中,室温下搅拌反应3-4h。旋转蒸发仪旋蒸除去溶剂,残渣溶解在乙酸乙酯中,以饱和碳酸氢钠和盐酸溶液洗涤后,硫酸钠干燥,过滤、蒸发,以二氯甲烷-甲醇柱层析纯化,得到抑制剂i-b-5目标产物67mg,产率56%。
109、
110、1h nmr (600 mhz, cdcl3) δ 8.56 – 8.43 (m, 1h), 8.06 – 7.81 (m, 4h),7.72 – 7.70 (m, 2h), 7.60 – 7.50 (m, 2h), 7.20 (d, j = 8.1 hz, 2h), 7.17 (d, j = 8.0 hz, 1h), 5.42 – 5.27 (m, 1h), 3.74 (s, 2h), 3.69 – 3.63 (m, 2h), 2.36(d, j = 4.9 hz, 3h), 2.20 (ddd, j = 21.1, 13.7, 7.6 hz, 2h), 2.14 – 2.07 (m,2h)。
111、13c nmr (151 mhz, cdcl3) δ 197.07, 167.34, 167.27, 142.12, 129.68,129.26, 129.20, 129.17, 128.80, 128.75, 128.47, 128.39, 128.08, 127.81,127.43, 127.11, 126.95, 59.02, 46.07, 29.70, 24.65, 22.69, 21.44。
112、ms esi ( m/z): calcd for c25h25n2o3 [m+h]+: 401.19,found 401.37。
113、实施例4:以4-硝基苯磺酰氯为原料合成抑制剂i-c-3
114、将4-硝基苯磺酰氯(5.0mmol)和甘氨酸乙酸乙酯盐酸盐(306mg,5.0mmol)、三乙胺(2.5ml,15mmol)加入二氯甲烷(20ml)中,室温下搅拌4h,用稀盐酸溶液洗涤,形成产物iii沉淀。
115、然后将氢氧化钠(5.0mmol)加入含有产物iii的甲醇(15.0ml)与水(15.0ml)的混合溶液中,搅拌反应16h,以旋转蒸发仪除去溶剂,将残余物重溶于水中,稀盐酸洗涤酸化,析出固体产物,低温过滤、干燥,制备得到产物iv。
116、
117、将l-脯氨酸(0.667g,5.79mmol)和五氯化磷(1.206g,5.79mmol)加入到含有二氯甲烷(17.0ml)的25ml茄形瓶中,低温反应器中0℃下搅拌反应2h。用旋转蒸发仪将溶剂蒸发除去后,室温下干燥1h。
118、残渣中加入溴化苄(0.990g)和氯化铝(2.316g,17.37mmol),于60℃加热反应4h,冷却至室温,倒入1m hcl溶液与碎冰的混合物中,分离出水相,以过量碳酸钠中和,加入适量二氯甲烷,过滤,少量二氯甲烷洗涤碳酸钠滤饼,合并有机相,旋转蒸发仪旋干后,经柱色谱分离,得到产物v。
119、
120、将产物iv(67mg,0.3mmol)溶于3ml dmf中,加入dipea(0.6ml,1.5mmol)和hatu(73mg,0.3mmol),15min后加入到含有产物v(80mg,0.3mmol)的dmf溶液(0.5mmol)中,室温下搅拌反应3-4h。旋转蒸发仪旋蒸除去溶剂,残渣溶解在乙酸乙酯中,以饱和碳酸氢钠和盐酸溶液洗涤后,硫酸钠干燥,过滤、蒸发,以二氯甲烷-甲醇柱层析纯化,得到抑制剂i-c-3目标产物61mg,产率43%。
121、
122、1h nmr (600 mhz, cdcl3) δ 8.37 – 8.14 (m, 3h), 7.79 – 7.34 (m, 2h),7.19 (dt, j = 47.2, 24.4 hz, 2h), 6.99 (s, 2h), 5.97 – 5.60 (m, 1h), 5.08 (s,2h), 4.07 (t, j = 6.8 hz, 2h), 3.82 – 3.70 (m, 2h), 2.33 – 2.30 (m, 2h), 2.06(s, 2h)。
123、13c nmr (151 mhz, cdcl3) δ 176.95, 174.42, 169.03, 144.59, 143.83,141.54, 139.02, 138.77, 131.30, 130.15, 128.59, 124.83, 60.99, 60.39, 55.56,34.78, 29.86, 29.70。
124、ms esi ( m/z): calcd for c21h21brn3o5 [m+h]+: 475.07,found 475.27。
125、实施例5:以4-甲氧基苯磺酰氯为原料合成抑制剂i-e-5
126、将4-甲氧基苯磺酰氯(5.0mmol)和甘氨酸乙酸乙酯盐酸盐(306mg,5.0mmol)、三乙胺(2.5ml,15mmol)加入二氯甲烷(20ml)中,室温下搅拌4h,用稀盐酸溶液洗涤,形成产物iii沉淀。
127、然后将氢氧化钠(5.0mmol)加入含有产物iii的甲醇(15.0ml)与水(15.0ml)的混合溶液中,搅拌反应16h,以旋转蒸发仪除去溶剂,将残余物重溶于水中,稀盐酸洗涤酸化,析出固体产物,低温过滤、干燥,制备得到产物iv。
128、
129、将l-脯氨酸(0.667g,5.79mmol)和五氯化磷(1.206g,5.79mmol)加入到含有二氯甲烷(17.0ml)的25ml茄形瓶中,低温反应器中0℃下搅拌反应2h。用旋转蒸发仪将溶剂蒸发除去后,室温下干燥1h。
130、残渣中加入萘酚(1.522g)和氯化铝(2.316g,17.37mmol),于60℃加热反应4h,冷却至室温,倒入1m hcl溶液与碎冰的混合物中,分离出水相,以过量碳酸钠中和,加入适量二氯甲烷,过滤,少量二氯甲烷洗涤碳酸钠滤饼,合并有机相,旋转蒸发仪旋干后,经柱色谱分离,得到产物v。
131、
132、将产物iv(63mg,0.3mmol)溶于3ml dmf中,加入dipea(0.6ml,1.5mmol)和hatu(73mg,0.3mmol),15min后加入到含有产物v(72mg,0.3mmol)的dmf溶液(0.5mmol)中,室温下搅拌反应3-4h。旋转蒸发仪旋蒸除去溶剂,残渣溶解在乙酸乙酯中,以饱和碳酸氢钠和盐酸溶液洗涤后,硫酸钠干燥,过滤、蒸发,以二氯甲烷-甲醇柱层析纯化,得到抑制剂i-e-5目标产物69mg,产率53%。
133、
134、1h nmr (600 mhz, cdcl3) δ 10.66 (s, 1h), 8.76 – 8.67 (m, 1h), 8.09 (t, j = 7.7 hz, 1h), 7.88 – 7.84 (m, 2h), 7.79 (d, j = 8.7 hz, 2h), 7.77 (s, 1h),7.19 (d, j = 9.0 hz, 1h), 7.15 (d, j = 9.0 hz, 1h), 6.92 (d, j = 8.8 hz, 2h),5.35 (t, j = 4.8 hz, 1h), 4.22 (d, j = 4.8 hz, 2h), 3.85 (d, j = 3.7 hz, 3h),3.46 (s, 2h), 2.21 (s, 2h), 2.01 (s, 2h)。
135、13c nmr (151 mhz, cdcl3) δ 191.84, 171.06, 155.96, 153.61, 151.79,131.53, 129.93, 129.73, 129.46, 129.31, 128.93, 128.57, 123.06, 114.35,113.82, 112.97, 63.63, 47.63, 46.89, 31.71, 29.69, 20.93。
136、ms esi ( m/z): calcd for c25h25n2o5 [m+h]+: 433.18,found 433.01。
137、实施例6:以喹啉-4-羧酸为原料合成抑制剂i-f-6
138、将喹啉-4-羧酸(5.0mmol)加入到二氯甲烷(20ml)中,滴加无水dmf(0.5ml)和草酰氯(5.0ml),室温下搅拌20h制备产物ii。
139、以旋转蒸发仪除去溶剂和多余的草酰氯,剩余残渣用二氯甲烷(20ml)溶解,加入甘氨酸乙酸乙酯盐酸盐(306mg,5.0mmol)和三乙胺(2.5ml,15mmol),室温下搅拌4h,用稀盐酸溶液洗涤,形成产物iii沉淀。
140、然后将氢氧化钠(5.0mmol)加入含有产物iii的甲醇(15.0ml)与水(15.0ml)的混合溶液中,搅拌反应16h,以旋转蒸发仪除去溶剂,将残余物重溶于水中,稀盐酸洗涤酸化,析出固体产物,低温过滤、干燥,制备得到产物iv。
141、
142、将l-脯氨酸(0.667g,5.79mmol)和五氯化磷(1.206g,5.79mmol)加入到含有二氯甲烷(17.0ml)的25ml茄形瓶中,低温反应器中0℃下搅拌反应2h。用旋转蒸发仪将溶剂蒸发除去后,室温下干燥1h。
143、残渣中加入2-萘酚(833mg)和氯化铝(2.316g,17.37mmol),于60℃加热反应4h,冷却至室温,倒入1m hcl溶液与碎冰的混合物中,分离出水相,以过量碳酸钠中和,加入适量二氯甲烷,过滤,少量二氯甲烷洗涤碳酸钠滤饼,合并有机相,旋转蒸发仪旋干后,经柱色谱分离,得到产物v。
144、
145、将产物iv(69mg,0.3mmol)溶于3ml dmf中,加入dipea(0.610ml,1.5mmol)和hatu(73mg,0.3mmol),15min后加入到含有产物v(72mg,0.3mmol)的dmf溶液(0.5mmol)中,室温下搅拌反应3-4h。旋转蒸发仪旋蒸除去溶剂,残渣溶解在乙酸乙酯中,以饱和碳酸氢钠和盐酸溶液洗涤后,硫酸钠干燥,过滤、蒸发,以二氯甲烷-甲醇柱层析纯化,得到抑制剂i-f-6目标产物63mg,产率46%。
146、
147、1h nmr (600 mhz, cdcl3) δ 8.71 (d, j = 8.2 hz, 2h), 8.07 (d, j = 9.0hz, 2h), 7.79 (d, j = 8.1 hz, 2h), 7.66 – 7.61 (m, 2h), 7.48 – 7.39 (m, 2h),7.14 (d, j = 9.0 hz, 2h), 5.46 – 5.20 (m, 1h), 4.22 (s, 2h), 2.17 – 2.14 (m,2h), 2.03 – 1.95 (m, 2h)。
148、13c nmr (151 mhz, cdcl3) δ 199.92, 173.41, 171.04, 140.68, 131.60,129.93, 129.49, 129.41, 129.26, 128.84, 128.67, 128.57, 126.50, 125.14,123.65, 123.07, 117.46, 114.36, 111.61, 107.06, 102.95, 63.64, 47.65, 37.43,29.70, 24.33。
149、ms esi ( m/z): calcd for c27h24n3o4 [m+h]+: 454.18,found 454.05。
150、实施例7:甘氨酸-吡咯烷类成纤维细胞活化蛋白抑制剂对成纤维细胞活化蛋白活性的抑制作用
151、以先导化合物compounds 4作为对照,试验选择u87mg细胞表达fap,以香豆素(amc)作为荧光基团,运用荧光探针suc-gly-pro-amc对fap体外酶活性进行检测,通过其水解产生amc的荧光响应反映fap的活性。
152、u87mg细胞由北京师范大学教育部重点实验室提供,suc-gly-pro-amc通过商购得到。
153、将u87mg细胞置于含1%(v/v)青霉素-链霉素和10%(v/v)胎牛血清的dmem培养基中,于37℃、5%co2培养箱中培养,待细胞融合度达80%-90%时,使用0.25%含edta胰蛋白酶溶液消化,传代培养,取对数生长期细胞用于试验。
154、将消化的u87mg细胞系吹匀为单个细胞的pbs混悬液,细胞计数100万个/ml。将本发明合成的fap抑制剂分别稀释成100pm、10nm、100nm、1μm、10μm共5个浓度梯度,将suc-gly-pro-amc稀释至25μm作为底物。
155、在96孔板上,每孔依次加入100μl细胞,50μl不同浓度的各抑制剂,50μl底物。同时设置对照组,只加入100μl细胞和100μl底物。
156、使用酶标仪在37℃进行检测,读取60min内激发波长380nm、发射波长460nm处的荧光强度,读取间隔为15min一次。
157、以时间为横坐标,荧光强度为纵坐标绘制曲线,在graphpad prism 9软件中进行非线性回归拟合得到各抑制剂的ic50值,结果列于表1中。
158、
159、表1中,作为阳性对照的先导化合物compounds 4对fap蛋白的半数抑制浓度为86.00nm,与其比较,本发明合成的大部分化合物均优于阳性对照,其中,化合物i-a-3、i-b-4、i-c-3、i-d-2、i-e-4、i-f-3对于fap均具有很强的抑制活性。
160、通过上述实施例证明,根据本发明所提供甘氨酸-吡咯烷类fap抑制剂对成纤维细胞活化蛋白具有很强的抑制剂作用,对比改造前的抑制剂先导化合物compounds 4,本发明提供的甘氨酸-吡咯烷类化合物的活性显著提高,可开发为新一代的抗癌药物。
161、本发明以上实施例并没有详尽叙述所有的细节,也不限制本发明仅为以上所述实施例。本领域普通技术人员在不脱离本发明原理和宗旨的情况下,针对这些实施例进行的各种变化、修改、替换和变型,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!