利用短MPS读数确定长DNA序列的制作方法
本技术涉及一种dna测序方法。
背景技术:
1、目前,dna测序还有改进的空间。
技术实现思路
1、在某些方面,本技术提供了一种dna测序方法,包括a)将长dna分子应用于索引阵列(indexed array),其中i)索引阵列包括转移位点(transfer site,ts)阵列,每个ts包括基底和连接在或位于基底上的克隆条形码(source of clonal barcode,scb)源,每个scb包括唯一可转移条形码序列的许多拷贝,且与每个ts相关的唯一可转移条形码序列是已知的,以及ii)应用于索引阵列的长dna分子在应用时或应用后处于拉长构象;b)在多个该ts中的每个ts上,将唯一可转移条形码序列从ts的scb部分启动转移到长dna分子上靠近ts的位置;c)从该索引阵列中回收长dna分子片段;d)对片段进行测序以产生序列读数,其中至少部分序列读数包括唯一条形码序列和来自长dna分子的序列;e)通过将读数中的所述唯一条形码序列与索引阵列中包含条形码的ts的位置相关联,对(d)中的序列读数进行排序,以及根据阵列中ts的位置的相对靠近性对读数排序。
2、定义
3、“单一反应混合物”中的组分或反应是指在标记步骤期间,在单一混合物中进行反应,不需要分隔成单独的试管、容器、等分液、孔、室或液滴。组分可同时或以任何顺序加入,以制成单一反应混合物。
4、术语“交错单链断裂(staggered single-stranded break)”指的是引入双链或部分双链dna分子的单链中的断裂(由切口或间隙产生),导致多个重叠的单链核酸片段与其他单链核酸片段杂交。其中至少有一些核酸片段,5′序列的一部分与另一个核酸片段的5′序列的至少一部分互补,并且3′序列的至少一部分与又另一个核酸片段的3′序列的至少一部分互补,这样在杂交条件下,多个核酸片段相互杂交形成核酸复合物。为了说明而非限制,图4展示了由交错单链断裂分离的四个核酸片段组成的核酸复合物。可以理解的是,核酸复合物(或“复合物”)可以且通常包含四个以上的核酸片段。
5、术语“部分双链”是指两条dna链相互杂交,其中一条链的至少一部分未与另一条链杂交。部分双链dna的两条dna链可以长度不同,也可以长度相同。
6、本文所用的“唯一分子标识符(umi)”是指存在于dna分子中的核苷酸序列,可用于区分单个dna分子。参见,例如,kivioja,nature methods 9,72-74(2012)。可以对umi及与识别来自同一源核酸的序列读数相关的dna序列进行测序。本文使用的术语“umi”既指umi的核苷酸序列,也指物理核苷酸,这一点从上下文中可以明显看出。umi可以是随机、伪随机,或部分随机,或非随机的核苷酸序列,它们被插入接头(adapter)或以其他方式结合到待测序的源核酸(如dna)分子中。在某些实施方案中,每个umi可唯一识别样本中存在的任何给定源dna分子。
7、如本技术所用,术语“单管lfr”或“stlfr”指的是例如美国专利公开2014/0323316和wang et al.,genome research,29:798-808(2019)中描述的过程,其全部内容在此作为参考全部并入本技术,其中,除其他外,相同的、唯一的条形码序列(或“标签”)的多个拷贝与单个长核酸片段相关联。在单管lfr的一个实施方案中,长核酸片段以一定的间隔用“插入寡核苷酸”标记。在一个实施方案中,插入寡核苷酸由一种或多种酶(如转座酶、切口酶和连接酶)引入长核酸分子。不同长核酸片段的条形码序列是不同的。因此,标记单个长核酸片段的过程可在例如单个容器中方便地进行,无需分隔。这一过程可对大量单个dna片段进行分析,而无需在标记步骤中将片段分离到单独的试管、容器、等分液、孔或液滴中。
8、本文中使用的“唯一”条形码是指与单个珠子(bead)相关并可用于区分单个珠子的核苷酸序列。在每个都具有唯一条形码的珠子群体中,与一个珠子相关的条形码序列至少与群体中90%珠子的条形码序列不同,更常见的是至少与群体中99%珠子的条形码序列不同,甚至更常见的是至少与群体中99.5%珠子的条形码序列不同,最常见的是至少与群体中99.9%珠子的条形码序列不同。
9、与多核苷酸和基底(例如珠子)相关的术语“连接”指的是多核苷酸(或多核苷酸的一个末端)直接与基底接触或共价连接。例如,表面可能具有与多核苷酸分子上的官能团反应形成共价连接的活性官能团。作为一个示例,通过将条形码寡核苷酸或杂交寡核苷酸连接到珠子上,将b-bla固定在珠子上。
10、当术语“溶液中”用于本技术公开的方法或组合物中使用的接头(或任何其他多核苷酸或多核苷酸复合物)时,是指接头(或任何其他多核苷酸或多核苷酸复合物)没有固定在基底上,可以在溶液中自由移动。当用于描述反应时,如″在溶液中进行的反应″是指反应发生在核酸之间,而所有核酸都在溶液中。
11、本技术中使用的术语“接头”有不同的含义,从上下文中可以明显看出。在某些实施方案中,“接头”指的是下文讨论的“分支连接接头(bla)”。在某些实施方案中,“接头”指的是下文讨论的“l-接头”。固定在珠子上的bla称为珠联分支连接接头(“b-bla”)。溶液中的bla称为溶液分支连接接头(“s-bla”)。
12、术语″接头的核酸片段″是指包含一个目标核酸片段和一个或多个接头序列的多核苷酸。例如,一个或多个接头序列可以是b-bla中的序列或l-adapter中的序列,或两者兼而有之。
13、术语“过量接头”(例如,过量的b-bla接头)或“未连接的接头”是指固定在珠子上的接头,尽管处于其他珠子接头与靶核酸片段连接的状态,但未与靶核酸片段连接。
14、术语“延伸的核酸片段”或“条形码延伸产物”是指与接头连接并延伸到包含条形码拷贝的片段。
15、术语“连接产物”或“连接接头”是指包含目标核酸片段和至少一个来自b-bla接头的接头序列的产物。在某些情况下,连接产物可进一步包括一端来自b-bla的接头序列和另一端来自另一接头(如l-接头)的接头序列。
16、术语“连接的第一接头”是指目的核酸片段与第一接头序列连接形成的产物。
17、术语“接头序列”指接头任一链上的序列,这一点从上下文中可以清楚地看出。也就是说,″接头序列″既可以指一条链上的接头序列,也可以指第二条链上的互补序列。例如,b-bla接头序列可以是条形码寡核苷酸上的序列,也可以是杂交寡核苷酸上的序列。
18、术语“分支连接接头”、“分支接头”或“bla”指的是部分双链接头。所述部分双链接头包括(i)双链平末端,包括一条链的5′末端和互补链的3′末端;(ii)单链区,包括条形码序列。如下文所述,分支接头双链区的5′末端可通过分支连接与核酸片段的3′末端连接。
19、术语“珠固定分支连接接头”或“b-bla”是指固定在珠子上的分支连接接头。本文公开的b-bla包括条形码寡核苷酸和杂交寡核苷酸,它们相互杂交。
20、术语“条形码寡核苷酸”是指包含条形码序列的b-bla链。
21、术语“杂交寡核苷酸”是指与条形码寡核苷酸互补的分支连接接头链。
22、术语“可逆终止子核苷酸”或“可逆终止子”是指具有3′可逆封端基团的核苷酸。“可逆阻断基团”是指可被裂解以在核苷酸的3′位提供羟基的基团,该羟基可与另一种核苷酸的5′磷酸基团连接。可逆封端基团可通过酶、化学反应、热和/或光来裂解。具有3′可逆封端基团的示例核苷酸在本领域是已知的,公开于美国专利号no.10,988,501,相关公开内容在此作为参考并入。
23、术语“复制”是指通过引物延伸产生模板的互补核苷酸链。
24、1.1概述
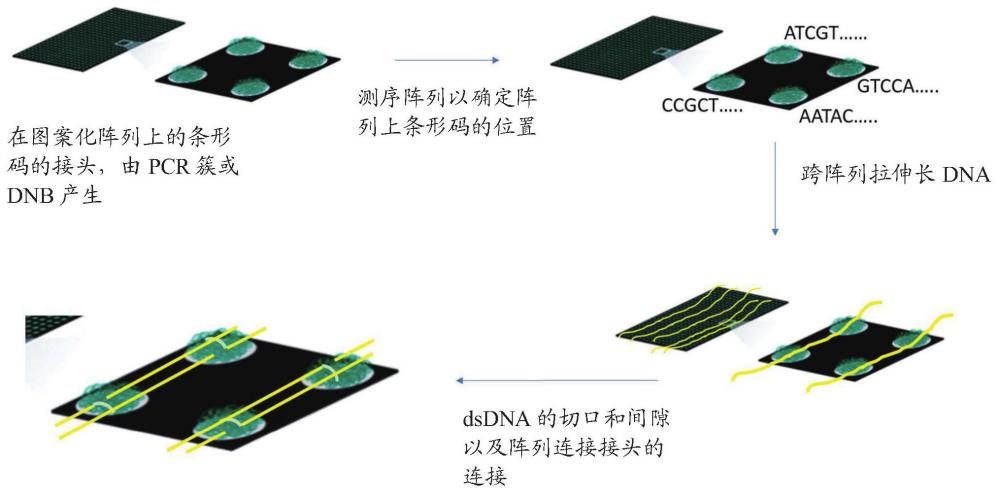
25、本发明用于利用短序列读数对长dna分子进行测序。每个短序列读数在原始长分子中的位置信息被保留下来。这对于正确组装来自具有高度重复dna碱基结构区域的短序列读数非常有用。图1提供了本发明的示例性工作流程。
26、在一种方法中,将感兴趣的延伸长dna分子应用到转移位点(ts)阵列上。见图2a,显示4x4阵列。每个单独转移位点的组成部分是(i)基底和(ii)克隆条形码源(scb)。如下所述,每个scb(因此,每个ts)由唯一的可转移条形码序列定义。
27、如图2a所示,当一个延伸的dna分子被应用到阵列中时,沿dna长度的不同区域靠近阵列中的不同转移位点(相应地,靠近不同的scb)。相应地,当长dna上的某个位置被定位为可以通过接收与scb关联的条形码来修改时,该位置是scb的近端。显而易见,延伸的长dna上靠近单个ts的位置也靠近ts的scb部分。图2a、2b和2c对此进行了示意性说明,其中编号位置为转移位点。
28、转移位点是阵列基底上的位置,scb在该位置上定位或固定。转移位点处的基底由物理特性定义,该物理特性将转移位点与基底周围的“非位点”区域区分开来。例如,转移位点的基底可能在化学上不同于非位点区域。例如,转移位点的基底可能具有吸引和保留scb的特性。其中一种方法是,转移位点基底具有亲水性,而非位点基底具有疏水性。一种方法是,转移位点具有磁性,而非位点没有磁性。在另一种方法中,转移位点包含结合条形码接头的捕获寡核苷酸,而非位点可能不包含捕获寡核苷酸。
29、在本公开的阵列中,转移位点通常以规则的模式排列,如行列、螺旋形、同心圆、蜂窝状、重复嵌套排列(如嵌套的“v”形)等。“线性”阵列只包括一行或两行。线性阵列通常与基底中的轨道或通道结合使用。这种轨道或通道可用于引导长dna分子,以改善和更好地控制dna分子的伸展和间距。在这种方法中,线性阵列的scb靠近dna。多个线性阵列(每个都与一个长dsdna相关)可以包含在同一个基底上或同一个流动池中。同样,除线性阵列外,还可将通道纳入阵列中。
30、scb和附属序列特征被配置为将条形码序列从ts转移到延伸的长dna分子中靠近ts及其相关scb的位置。见图2b,左下方,图2c。当scb与长dna分子上的位置足够接近从而条形码序列可以从scb转移到长dna上时,scb位于平铺在阵列上的长dna分子的近端位置。通过这种方式,可以用条形码序列沿长dna分子的长度对置于或者平铺在阵列的长dna分子进行标记,长dna中的标记位置与阵列中ts特异性条形码的位置相对应。术语“标签”、“标记”等具有本领域的通常含义,指条形码序列与长dna序列相关联。条形码序列可以与长dna序列毗连,或包含在与长dna序列毗连的条形码接头序列中,或以其他方式与长dna的区域相关联。
31、标记后,长dna从阵列中回收,分割并测序。可以理解的是,长dna并不是完整地回收;而是回收片段、扩增子、互补序列等,并进行测序,以产生测序“读数”。mps中的典型读数长度在200到1000个碱基之间。有些读数包含长dna序列和条形码序列。见图2b,左下方。由于条形码的位置与阵列上的ts位置相关,因此可以将短读数dna序列分配到dna分子长度方向上的位置。见图2b右下方。图2c表明,使用这种方法,两个相同的polya序列可以根据它们被分配到长dna的不同位置而区分开来。
32、引入的条形码之间的间距(相对于原始dna)部分受阵列上转移位点间距的控制。中心到中心的距离通常在200-500nm(600-1500碱基对)之间。在某些情况下,其范围为100-1000nm甚至更大。
33、1.1图1和图2的描述
34、图1显示了本文所述工艺的示例性工作流程。
35、图2a根据一种方法概述了本公开的各个方面。
36、图2b展示了带有36个转移位点(左上)的加载阵列,在该阵列上应用了单个长dsdna(右上)。制备和测序包含转移到原始长dna上(左下)的目标序列和条形码片段的测序文库。根据转移位点的已知位置(右下)确定原始长dna中读数(片段)的相对位置。
37、图2c是一幅漫画,说明在对高度重复的dna碱基序列进行测序时,如何利用位置信息来避免歧义。图中dna位于包含转移位点1-8的衬里阵列上方。在该图中,有两个区域的[a]11条带无法区分。利用本发明,“条形码2”从转移位点2转移到靠近一个[a]11的dna上,“条形码7”从转移位点7转移到靠近另一个[a]11的dna上。当对dna进行测序时,可以将包含[a]11和条形码2的读数与包含[a]11和条形码7的读数区分开来。
38、图3-13将在下文第1.9节中介绍。
39、1.3转移位点(包括克隆条形码源)及其阵列的配置
40、包括克隆条形码源部分在内的转移位点可以多种方式配置。一般来说,scb包括含有可转移条形码序列的条形码接头。在此上下文中,“条形码”具有本领域的通常含义,“可转移条形码序列”是指可从scb转移到本文所述长dna的条形码序列,以及″条形码接头″是指包含可转移条形码序列和下文所述附属序列特征的多核苷酸序列。
41、在本公开中,“条形码”和“条形码序列”可以互换使用。本文所用的条形码、条形码序列或条形码接头的转移可指寡核苷酸的物理转移和/或序列信息的转移。条形码序列的转移可指条形码序列本身的转移或条形码序列补码的转移。
42、1.3.1 scb格式
43、单个scb包含许多相同条形码序列的拷贝(即克隆群体)。除了识别特定转移位点多核苷酸克隆群体的唯一条形码外,scb通常还包括阵列上所有scb共享的辅助序列特征。辅助序列特征的例子包括:
44、(i)将条形码从scb转移到上覆长dna所需的序列元件,如转座子、杂交序列和3′支链连接(3′-bl)元件;
45、(ii)用于确定阵列上每个位置的条形码序列的序列元件,如引物结合位点(如用于测序和/或扩增)和/或锚定探针结合位点(如见us 9,267,172);
46、(iii)用于从标记的长dna片段制备文库的序列元件,包括接头、索引序列和引物结合位点;
47、(iv)扩增(例如rca或桥接扩增)所需的序列元件;
48、(v)在转移位点固定scb的序列元件;以及
49、(vi)用于实施本文公开的实施方案的其他序列元件。
50、为方便起见,我们将转移至长dna的序列称为“条形码接头”。条形码接头包含唯一的条形码以及附属序列特征。在某些出版物中,条形码接头被称为“条形码盒”。
51、现有技术中描述了scb(尽管本公开不限于先前描述的scb形式)。已经描述了许多scb。然而,它们是在用相同条形码的多个拷贝标记单个长dna的背景下描述的。同样,也有一些测序平台使用有序阵列,如下所述。可以对现有技术的scb和阵列进行修改,或将其并入本公开所述的新型创新方法和装置中。
52、1.3.1.1基于珠子的scb
53、在一种方法中,scb是被含有条形码接头序列的可转移条形码结合、被其覆盖或含有的珠子。在这种方法中,每个scb通常与含有相同可转移条形码的短多核苷酸的多个拷贝--至少1000个、至少10,000个、至少100,000个或至少1,000,000个--相关联。也就是说,每个scb都与条形码的克隆群体相关联。
54、在一种基于珠子的方法中,转移位点可以是纳米孔,其中许多或大多数纳米孔被单个珠子(因此是单个条形码克隆群)占据。在这种方法中,单个孔的大小可以接受单个珠子,或者,珠子的大小可以使单个珠子占据一个孔,并排除其他珠子占据同一孔。珠子的一部分可以从孔的顶部(开口部分)突出,或者珠子可以通过其他方式接触到(靠近)覆盖的长dna。这种结构可称为“纳米孔阵列”。
55、在一种相关的基于珠子的方法中,scb是珠子,而转移位点可以是基底上离散的间隔位点,这些位点(相对于基底的其他区域)对scb具有亲和力。scb被基底上与scb没有亲和力的区域隔开。这种配置可称为“图案化阵列”。例如,在某些实施方案中,图案化阵列基底可以是基本平面、凹面、凸面或圆柱形。
56、1.3.1.2 dna基于纳米球的scbs
57、在另一种方法中,scb是dna纳米球(dnb),其中的单体包括条形码接头。转移位点可以是由单个dnb占有的孔。单个孔的大小可以接受单个dnb,或者,dnb的大小可以占据单个孔,并排除其他dnb占据同一孔。dnb的一部分可以从孔的顶部(开放部分)突出,或者dnb可以以其他方式与覆盖的长dna相接触。
58、在一种相关的方法中,scb是dnb,并且转移位点可以是基底上与dnb有亲和力的离散间隔区域,与dnb没有亲和力的基底区域隔开。drmanac et al.“human genomesequencing using unchained base reads on self-assembling dna nanoarrays.”science 327.5961,中描述了示例阵列。
59、1.3.1.3基于群集的scbs
60、在一种方法中,scb是基底上离散的、间隔开的扩增子克隆簇。每个簇中的扩增子包括簇特异性条形码序列。扩增子簇可以放置在基本平面、凹面、凸面或圆柱形的基底上,也可以放置在带有孔(如浅孔)的基底上。簇的位置或配置允许条形码序列转移到长dna的近端位置。
61、1.3.2随机阵列
62、本文所述阵列为“随机阵列”。珠子、dnb和扩增子在阵列上随机分布,因此在对单个阵列进行解码之前,任何给定ts中scb所代表的条形码的身份(序列)都是未知的。见图1。
63、珠子阵列可通过在纳米孔阵列基底或图案化阵列基底上分布珠子来制备,由于尺寸排斥、亲和性或其他机制,通常每个纳米孔或离散间隔位点只捕获单个scb。
64、dnb阵列可通过在纳米孔阵列基底或图案化阵列基底上分布dnb来制备,由于尺寸排斥、亲和性或其他机制,通常每个纳米孔或离散间隔的位点只能捕获单个scb。
65、簇阵列可通过将scb模板分配到纳米孔或离散间隔位点,然后使用桥式pcr或类似方法进行扩增而形成。机械和/或动力学方法(包括排除扩增)可用于确保簇是克隆的(例如,共享相同的条形码)。参见如,wo2013/188582a1,通过引用并入本文。
66、基本(未解码)阵列可在使用前预先制作,并以试剂盒形式提供,无需解码。
67、1.4对数组解码以产生索引数组
68、基本阵列制备完成后,确定阵列上每个ts位置的条形码序列,从而生成“索引阵列”。每个ts上的条形码序列可使用标准测序方法确定。本领域技术人员会认识到,本公开内容中使用的阵列可能与广泛使用的测序阵列相同,或者是对其进行了修改,这样在解码步骤中就可以使用已知的协议和市售试剂。例如,可以使用通用产品(有时经过适当修改)来确定阵列上的条形码序列,包括但不限于mgi tech平台(通常是基于dnb的阵列,例如使用mgiseq-2000 fcl)、illumina(基于簇的阵列)和ion torrent平台(基于珠的阵列)。合适的方法包括使用可逆终止核苷酸的合成测序(sbs)、杂交测序(sbh)或cpal(见drmanac etal.2002,“sequencing by hybridization(sbh):advantages,achievements,andopportunities”in advances in biochemical engineering/biotechnology,vol.77;uspat.no.8,518,640),以及离子半导体测序(见merriman et al.,2012,electrophoresis33.23:3397-3417)。
69、当每个转移位点的条形码序列已知时(即与阵列上每个地址相关的特定条形码序列已知),阵列可称为″索引阵列″。索引阵列可以在使用前制作,也可以成套提供。
70、1.5将一个或多个长dna分子应用于索引阵列以产生“加载阵列”
71、将一个或多个长dna分子应用于索引阵列。应用了长dna的索引阵列可称为“加载阵列”。
72、长dna片段可以从任何具有基因组dna的生物体中获得。长dna的长度可以从1kb到500kb或更长。长dna一般长于10kb,长度可达到一个或多个兆位。在某些实施方案中,长dna是基因组dna。在实施例中,长dsdna可以是10-100kb、10-500kb、20-300kb、50-200kb、100-400kb、100kb至1mb或100kb至10mb。
73、长dna分子可以用已知的技术方法获得。参见,例如,sambrook and russell,molecular cloning:a laboratory manual,third edition,cold spring harbor press;peters et al.,nature 487:190-195(2012)。本文所述的方法可用于不同大小的dna分子(如10kb至数百万个碱基)。
74、应用于阵列的长dna通常为双链(“长dsdna”)。然而,在某些实施方案中也使用单链长dna。在某些方法中,条形码被插入单链dna中。在某些方法中,单链dna在阵列上被制成双链或部分双链。在一种方法中,3′端含有一定数量随机碱基的条形码延伸引物可与单链dna杂交,然后进行延伸反应以复制dna。
75、1.5.1长dna的预处理
76、在某些情况下,长dna会在下面描述的标记步骤之前进行处理。例如,一种方法是通过转座将杂交序列插入长dna中。整合的杂交序列可作为scb上互补捕获序列的陷阱。在某些方法中,杂交序列平均每200-1000个碱基对插入长dna分子中。
77、例如但不限于此,wang等人描述了利用转座法对长双链dna进行预处理,以每隔一定时间插入杂交序列。参见wang et al.“efficient and unique cobarcoding ofsecond-generation sequencing reads from long dna molecules enabling cost-effective and accurate sequencing,haplotyping,and de novo assembly.”genomeresearch 29.5(2019),并入本文作为参考。然后,插入的杂交序列可用作“陷阱”,捕获显示在珠子表面的条形码接头的互补单链区域。在一种方法中,dna序列被tn5转座酶结合,其中包含用于杂交的单链区和被转座酶识别并实现转座反应的双链序列。
78、在某些方法中,将预处理的长dna(例如,含有通过转座插入的转座子杂交序列的dna)应用于转移位点阵列以进行定位标记。在某些方法中,长dna在应用到阵列后进行“原位”处理。在某些方法中,长dna在应用于阵列之前和之后都要经过处理。
79、1.5.2将长dna应用于阵列
80、将长dna分子应用于索引阵列时,最好将其拉伸或拉长。在这里,“拉长”(或等同于“延长”或“拉伸”)是指dna的不同部分靠近不同的转移位点,一般来说,dna分子中相距较远的序列往往与阵列上相距较远的转移位点相关联。完全伸长的dna的理论长度约为每个碱基对0.34nm。就本公开而言,如果分子(或分子在阵列上的部分)的端到端长度至少是理论最大长度的约50%、约75%或约80%,则阵列上的单个分子是拉长的。单个分子的拉长部分(“部分”由两个边界定义)的两个边界之间的距离至少为理论最大值的50%、75%或80%。可以认识到,在某些情况下,阵列上的长dna分子只有一部分可能被拉长(“拉长部分”),并可用于根据本公开内容进行测序。可以使用多种方法将长dna分子应用到阵列上。例如,美国专利号no.8,153,438(″sequencing nucleic acid polymers with electronmicroscopy")描述了使用针型工具在基底上延伸多核苷酸链的方法。在某些情况下,使用“分子梳理”(例如,见本文引用的wo2010/115122a2;另见chan et al.,2006,“a simpledna stretching method for fluorescence imaging of single dna molecules”nucleic acids research 34.17:e113-e113(本文引用并入),以及本文引用的参考文献)或其他方法拉长dna分子。另见lim et al.,2001,genome research 11.9(2001):1584-1593。在某些方法中,单个dna分子通过流体在基底上的流动而被拉长或“拉伸”。在某些情况下,dna在流动过程中被固定。如上文第1.1节所述,在某些情况下,阵列包含用于延伸长dna分子的通道(如沟槽或轨道)。
81、应用于阵列的长dna可在原位进行修饰(如片段化、双链化或扩增)。一旦应用到阵列上,避免片段化并不重要(尽管避免过度片段化可能对某些应用或文库构建有用)。在某些情况下,长dna在预处理过程中已被片段化,但这些片段被一种保持连续性的转座酶固定在一起(见amini et al.,2014,nat.genet 4 6,1343-1349)。这种片段长dna的应用被视为单个长dna分子的应用。
82、在下面的描述中,加载阵列通常被描述为单个长dna已应用到阵列中。这只是为了便于描述。可以理解的是,多个(如2、3、4、5、6或6个以上)dna分子可应用于一个阵列。
83、1.6标记长dna
84、条形码序列从转移位点的scb转移到铺设在ts上的长dna分子上。这种转移可称为“标记”长dna或其片段、扩增子或其互补序列。
85、已经描述了多种标记长dna的策略。这些策略包括基于转座子的策略和标记策略,下文将举例说明。在本公开内容的指导下,本领域技术人员将了解如何修改这些先前描述的方法,以用于本文所述的基于阵列的标记。
86、1.6.1基于转座子的方法
87、使用珠子或dnb的基于转座子的标记方法已经被描述过。参见wang et al.“efficient and unique cobarcoding of second-generation sequencing reads fromlong dna molecules enabling cost-effective and accurate sequencing,haplotyping,and de novo assembly.”genome research 29.5(2019):798-808;drmanacwo 2014/145820“multiple tagging of long dna fragments”;peters etal.wo2020157684a1“high coverage stlfr”;zhang et al.,haplotype phasing ofwhole human genomes using bead-based barcode partitioning in a singletube.nat biotechnol 35,852-857(2017);gormley et al.wo2014/108810a2“samplepreparation on a solid support.”。为所有目的,上述各项均并入本文作为参考。在某些基于溶液的标记方法中,dnb或珠子各自显示100,000个或更多个相同的唯一条形码,用于标记具有多份相同条形码的单个dna分子。含有特定条形码的读数可被分配给相同的dna分子。相反,在本方法中,用不同的条形码标记单个dna分子,带有特定条形码的读数可被分配到长dna的某个位置。
88、1.6.2基于切口连接的方法
89、基于连接的标记方法用于标记长dna已有描述。参见,drmanac wo 2014/145820“multiple tagging of long dna fragments”;peters wo2019217452a1“single tubebead-based dna co-barcoding for accurate and cost-effective sequencing,haplotyping,and assembly”;peters et al.wo2020157684a1“high coverage stlfr”和构成了下文第1.9节(“切口连接stlfr”)的基础的题为“nick-ligate stlfr”的pct/cn2022/107241。为所有目的,上述各项内容均以引用方式并入本文。广义上讲,这种方法是在双链dna中引入一个切口或间隙,然后将条形码接头连接(有时使用3′分支连接)到切口或间隙处产生的游离端。切口连接法通常利用3′分支连接反应的特性。3′分支连接描述见wang et al.,“3’branch ligation:a novel method to ligate non-complementary dnato recessed or internal 3’oh ends in dna or rna”biorxiv,june 29,2018,doi:https//doi.org/10.1101/357863。通过将接头的″长″臂结合到连接到珠子或基底上的序列中,然后将构建体暴露于接头的″短″臂,可以在scb上构建多个3′分支连接接头(3′-bla")。然后,短臂可与长臂退火,产生3′_bla的特征性″y″构型。
90、1.6.3 dnb方法
91、wo 2014145820a2中一般描述了使用dnb作为scb进行标记的方法,本文并入作为参考。在一种方法中,用切口酶或限制性内切酶分割带有多份条形码接头和与杂交序列互补的区域(通过转座或切口连接引入)的dnb,以产生可用于转移到上覆dna的游离末端。在一种方法中,寡核苷酸与条形码相邻杂交,并带有5′或3′非杂交尾部。尾部可以是5′po4或3′oh或被阻断的3′(带有双脱氧核苷酸或其他阻断基团)。在5′尾部的情况下,条形码序列是通过引物延伸从3′杂交末端通过条形码序列复制的。然后,5′po4尾部可与3′阻断寡聚物杂交,并可用于3′分支连接。或者,如果3′侧被用作尾部,那么可以从条形码5′侧的额外引物进行延伸,然后与3′尾部寡聚物的5′侧连接。然后,该寡聚物可与另一个寡聚物杂交,形成一个dsdna区域,该区域将成为转座酶的结合位点,并可通过转座反应插入目标dna中。
92、1.6.4集群的使用
93、用于dnb的策略也可用于基因簇。此外,还可通过桥式pcr或类似的扩增策略将连接或转座所需的序列添加到簇分子的末端。然后,可将簇变性,并与寡核苷酸杂交,形成用于转座或3′分支连接的dsdna区域,如dnb所述。
94、1.6.5启动标记
95、将长dna应用于阵列后,开始转座过程。启动可以通过多种方式完成。例如,可以通过向阵列中注入转移所需的试剂(如酶(聚合酶、切酶、连接酶、转座酶)或辅助因子)和/或通过改变ph值和温度等条件来促进转移。
96、1.7.释放标记的长dna并制备测序文库
97、转移条形码后,释放或回收标记片段。捕获过程取决于转移方法和操作者的喜好。在一种释放方法中,通过改变温度或离子条件(释放与scb序列退火但未共价连接的片段)将长dsdna的标记部分从珠子上去除。在一种方法中,长dsdna的标记部分是通过酶解或化学裂解连接序列来释放的。释放的片段可在释放后进行扩增和/或通过添加接头进行修饰。
98、在一种优选方法中,长dna的标记部分通过扩增释放,其中标记过程中引入的序列包含扩增引物结合位点。扩增子包含标记序列和基因组序列。应该认识到,扩增释放本身并不导致物理的盒的释放,而是导致扩增子的释放。
99、可从标记片段中制备测序文库,并使用常规方法确定序列。确定的读数包括条形码序列以及目标(如基因组)序列。
100、1.8.排列读数
101、如图2b和2c所示,包含基因组序列的读数根据条形码序列与阵列上位置之间的对应关系进行组合。例如,两个条形码(即两个含条形码的ts)之间的距离和方向是已知的,来自这些已知条形码的读数可利用此信息从5′到3′定向。此外,在大多数情况下,一些读数会映射到参考基因组。这样就可以将一组读数置于基因组中相对较小的部分。通常会对具有相同基因组区域的多个长dna进行测序,这样第二个分子的读数就可以相对于第一个分子交错放置在相同区域,从而产生重叠覆盖。
102、1.9.用于切口连接stlfr的方法
103、以下公开的内容基于待决未公开专利申请pct/cn2022/107241,其内容通过引用并入本文。
104、在一个方面,本公开提供了一种制备用于测序的接头多核苷酸文库的方法,包括在单个反应混合物中a)将双链目标核酸与一种或多种核酸切口剂接触,以产生由交错的单链断裂分隔的多个重叠核酸片段;(b)提供多个珠子,每个珠子包括固定在珠子上的多个分支连接接头(b-bla),并提供在3′末端具有简并序列的l-接头群体;以及(c)在连接酶存在的情况下,将b-bla与至少一种核酸片段接触、(d)在有连接酶存在的情况下,将l-接头群体与核酸片段的至少一个接触,从而将l-接头连接到核酸片段的5′末端,由此获得在5′末端具有l-接头序列、在3′末端具有b-bla接头序列的核酸片段文库。
105、在另一方面,本文公开了一种用于制备测序用多核苷酸文库的方法,该方法包括在单个反应混合物中:
106、(a)将双链目标核酸与一种或多种切口剂接触,以产生被交错的单链断裂分开的重叠核酸片段;以及
107、(b)在连接酶存在的情况下,将包含多个部分双链第一接头的珠子与核酸片段接触,其中每个第一接头包括:(i)双链平末端,包括一条链的5′末端和互补链的3′末端;(ii)固定在珠子上的单链区,其中单链区包括条形码、从而使用dna连接酶将至少一个第一接头的双链平末端中的链的5′末端与至少一个核酸片段的3′末端连接,以产生已连接的第一接头,其中已连接的第一接头包括条形码和至少一个核酸片段、(c)使连接的第一接头变性,以及(d)对与连接的第一接头中条形码3′相对序列杂交的引物进行受控延伸,从而产生与连接的第一接头互补的部分延伸链。
108、在另一个方面,本文公开了一种反应混合物,该混合物包含(1)一种或多种切口剂,(2)一种或多种连接酶,(3)由交错单链断裂分开的多个重叠核酸片段、(4)部分双链分支接头,包括条形码寡核苷酸和杂交寡核苷酸,彼此杂交以形成部分双链核酸分子,其中条形码寡核苷酸与珠子连接并包括条形码、其中杂交寡核苷酸不与珠子连接,其中部分双链核酸分子包括(i)具有5′末端和3′末端的双链平末端和(ii)包含条形码并具有单链末端的单链区,其中双链平末端的5′末端与至少一个核酸片段的3′末端连接。
109、图3显示了文库制备方法的示例性工作流程。
110、图4展示了对双链目标核酸(210)进行裂解以产生交错的单链断裂(220)。图2还说明了延伸断裂以创造性地延伸由断裂分隔的片段(240)之间的间隙(230),为连接接头做准备。
111、图5a和5b显示了在单个反应混合物中通过分支连接(320)将b-bla接头(320)添加到目的dna(310)的3′端并将l-接头(340)添加到目的dna的5′端的示例性方法。图3a显示,珠子(300)由固定在其上的b-bla组成。每个b-bla由两条链组成:1)条形码寡核苷酸,包括条形码序列(330)和3′端的双脱氧阻遏核苷酸;2)杂交寡核苷酸,与条形码寡核苷酸杂交。条形码寡核苷酸的5′端与珠子(300)连接。虽然为了更好地说明和解释,图中的步骤是分开的,但b-bla接头和l-接头的添加可以在一个反应中完成。b-bla接头(340)的条形码(330)通过延长未与珠子连接的链(350)进行复制,从而产生延长的核酸片段(360)。多余的b-bla接头(370)(即未连接到片段上的b-bla接头)也将被延长。延伸的核酸片段(360)可以使用两个引物退火至b-bla接头序列和l-接头序列的两个末端进行扩增。或者,如下文所述,可使用与两个接头序列退火的分离寡聚物对延伸核酸片段(360)进行环化。参见第10节″扩增″。多余的接头(370)没有l-接头,因此不能通过pcr扩增或环化。
112、图6显示了在一次反应中将b-bla(410)添加到目标dna的3′,将l-接头(420)添加到5′的示例性方法。每个b-bla的条形码寡核苷酸在3端被阻断(例如,通过双脱氧阻断核苷酸)。同一b-bla的杂交寡核苷酸可通过3′分支连接到目标核酸片段。将杂交寡核苷酸和目标核酸片段连接形成的连接产物(450)进行延伸,以结合来自b-bla的条形码(430),形成延伸核酸片段(460)。延伸核酸片段(460)可通过变性从珠子中释放出来,释放出来的片段再通过pcr扩增或环化。可选择用λ外切酶和外切酶降解多余的b-bla(440),以避免扩增未连接的接头。
113、图7显示了与图6类似的另一种示例方法,即在单个反应中将b-bla(510)添加到目标dna的3′,并将l-接头(520)添加到5′。b-bla被固定在珠子(500)上。在图6中,条形码寡核苷酸被阻止延伸,而在图7中,杂交寡核苷酸被阻止,条形码寡核苷酸可以连接到目标核酸片段上,产生条形码核酸片段(550);不需要通过延伸复制条形码。然后,过量的b-bla(560)和连接产物都会变性,从而产生单链条形码核酸片段(530),它仍然与珠子连接在一起。在一种方法中,b-bla包含条形码寡核苷酸3′端附近的尿嘧啶;如上所述产生的条形码核酸片段(530)可通过接触user从珠上释放。然后可以对释放的链(540)进行扩增或直接环化。也可以通过recj或exo7处理去除多余的b-bla(570)。“*”代表硫代磷酸键。
114、图8显示了本公开的一个说明性实施方案,其中b-bla在切口酶处理过程中与靶dna接触。与图6类似,每个b-bla的条形码寡核苷酸被阻断延伸;但在图8中,每个条形码寡核苷酸还可以在条形码序列(620)和双脱氧阻断核苷酸之间包含一个或多个尿嘧啶(610)。杂交寡核苷酸(630)可通过分支连接与目标核酸片段连接。然后加入user来裂解条形码寡核苷酸并释放双脱氧阻遏核苷酸,从而使条形码寡核苷酸具有可延伸的末端(650)。连接产物(630)被延伸以结合条形码,形成条形码核酸片段(640)。条形码寡核苷酸的3′端(650)不含阻遏核苷酸,也会被延伸。然后加入具有3′→5′外切酶活性的exoiii,完全降解多余的b-bla(660),并从3′→5′方向部分降解条形码核酸片段,从而得到部分杂交的条形码目标核酸片段(670)。然后,所述部分杂交的条形码目标核酸片段(670)被延伸形成双链条形码核酸片段(680),然后通过平末端连接与第二接头连接。在某些情况下,第二接头不含5′磷酸基团,以减少自连接。连接产物经变性后形成单链核酸片段(690),它的两端现在都有接头序列。单链核酸片段(690)现在可以通过pcr扩增或环化。
115、图9a和9b显示了本公开的另一个实施方案,其中b-bla被固定到珠子(700)上。每个b-bla包括相互杂交的条形码寡核苷酸(710)和杂交寡核苷酸(720)。杂交寡核苷酸在3′端包括双脱氧阻遏核苷酸,而条形码寡核苷酸在与条形码序列(790)相连的5′位点上包括尿嘧啶。图9a和9b展示了以下事件:1)条形码寡核苷酸与经过切口酶处理的靶核酸片段连接,通过分支连接形成条形码核酸片段(730)。2)通过变性去除杂交寡核苷酸;3)加入核酸酶,如recj或exovii,以降解单链过剩的b-bla(740);4)引物(750)退火至条形码核酸片段(730)上条形码5′的序列,并延伸形成双链dna分子(760);然后将双链dna分子连接到第二个双链接头(770)上,形成双链分子(780),其两端都具有接头序列,其中一个接头序列来自分支接头,另一个接头序列来自第二个双链接头。可以选择第二接头不包含5′磷酸,以避免自连接。然后,带有双接头序列的双链分子(780)会变性,并通过user从珠子中释放出来,形成单链分子(781),然后可以对其进行扩增和/或环化。
116、图10显示了本公开的一个说明性实施方案,其中b-bla被固定到珠子(800)上。每个b-bla包括条形码寡核苷酸(820)和杂交寡核苷酸(810)。条形码寡核苷酸(820)的3′端包含双脱氧阻遏核苷酸。在切口酶处理过程中,b-bla中的杂交寡核苷酸通过分支连接到目标核酸片段。在反应中加入λ外切酶和外切酶i,以去除多余的b-bla(830)。将杂交寡核苷酸和目标核酸片段连接形成的连接产物延伸复制条形码,从而得到条形码核酸片段(840),通过变性将其从条形码寡核苷酸中分离出来。引物退火到单链分子与条形码序列3′的序列上并延伸。延伸后形成双链分子(850),然后与第二个接头连接,形成双链核酸片段(860),其两端具有接头序列。然后可以通过pcr扩增双链核酸片段(860)。或者,也可以将双链核酸片段变性,形成单链核酸片段,然后将其环化。可选地,第二接头缺少5′磷酸,这可以最大限度地减少单个第二接头的自连接。
117、图11a和图11b显示了本公开的另一个实施方案,其中b-bla被固定到珠子(900)上。每个b-bla包括相互杂交的条形码寡核苷酸(910)和杂交寡核苷酸(920)。杂交寡核苷酸(920)的3′端包含双脱氧阻遏核苷酸。首先,条形码寡核苷酸与经过切口酶处理的靶核酸片段连接,通过分支连接形成条形码核酸片段(930)。其次,通过变性去除杂交寡核苷酸。第三,进行受控聚合酶延伸,留下可用于3′分支连接的5′悬空(940)。受控延伸只进行100-150个碱基,由不具有3-5′外切酶活性的dna聚合酶进行,从而在模板末端产生a尾(950)。这将导致多余接头的完全延伸和a尾,但连接到基因组片段的接头将是不完全的。接下来,与发夹式接头进行连接,发夹式接头的t尾与延伸的过量接头的a尾互补,从而阻止过量接头(960)的连接或延伸,而与目标核酸片段(970)连接的剩余接头不会被阻止(即这些剩余接头无法与发夹式接头连接)。终止子可以在不同的循环中以不同的浓度或在不同的时间点添加,以产生不同长度的延伸产物,从而在测序过程中提供每个片段大部分碱基的重叠覆盖。
118、剩余的接头(970)用可逆终止子进一步延伸,然后进行反应去除终止子阻断基团,再进行3′分支连接,将第二个接头(980)添加到末端的靶核酸片段上。然后将反应变性,再通过pcr扩增或环化得到两端包含两个接头序列的单链分子(990)。
119、图12a和图12b显示了本公开内容的另一个实施方案,其中涉及进行受控延伸。与图11a和图11b类似。本实施例中使用的b-bla也是分支接头,它包括相互杂交的条形码寡核苷酸和杂交寡核苷酸(1020)。杂交寡核苷酸(1020)的3′端包含双脱氧阻遏核苷酸。首先,条形码寡核苷酸与经过切口酶处理的靶核酸片段连接,通过分支连接形成条形码核酸片段(1030)。其次,通过变性去除杂交寡核苷酸。第三,使用具有3-5′外切酶活性的聚合酶进行受控聚合酶延伸,条件是将延伸限制在约100-150个碱基。这样就留下了可用于3′分支连接的5′悬空(1040)。这导致与目标核酸片段(1040)连接的接头延伸不完全,而多余的接头(1050)延伸完全。然后在反应中加入λ外切酶,λ外切酶降解带有5′磷酸的平端dsdna接头(1050)。与单链dna相比,λ外切酶更喜欢磷酸化的双链dna,因此接头的短插入物(如1050)比长dna插入物(如1040)更容易降解。如图12b所示,该方法的其余步骤与图11a和11b所示类似。
120、图13a显示了进行图12a中所述的受控延伸,完全延伸多余的接头(1150),部分延伸连接产物(1140)。图13b显示,部分延伸的连接产物(1140)在可逆终止子存在的情况下进一步延伸,然后去除可逆终止子中的终止子阻断基团,再与第二个接头(1160)连接。这样,多余的接头(1150)平端连接,条形码目标核酸片段(1170)的3′支连接,形成两端都有接头序列的核酸片段(1180)。未连接链(1190)由链置换聚合酶在延伸控制条件下延伸,使未连接链仅延伸约100-150个碱基。这种延伸导致固定在珠上的接头核酸片段发生链置换,并释放出接头核酸片段(1190)。释放出的接头核酸片段可以收集到溶液中。珠子可重复用于下一周期的受控延伸。与上述涉及可逆终止子的其他实施方案类似,终止子可以在不同的周期中以不同的浓度或在不同的时间点添加,以产生不同长度的延伸产物。这样做的好处是,在测序过程中,每个片段的大部分碱基都有重叠覆盖。
121、切口连接概述
122、本文描述了用于制备测序文库的″切口连接″或″切口联结″单管lfr方法。这些方法以可控速度、频率或两者兼有的方式在双链目标核酸中引入单链断裂(如切口或间隙)。如下文所述,这些方法还可将接头连接到断裂的3′(3-prime)侧、断裂的5′(5-prime)侧或切口或间隙的两侧。添加一个或多个接头可产生″接头片段″。制备文库所涉及的酶促反应,例如切接和连接,可以在单一混合物中进行,以产生带有所需接头和条形码的靶核酸文库。
123、切口连接方法具有某些优势,特别适用于大基因组片段测序序列读数的从头组装。
124、首先,该方法可产生重叠的单链核酸片段,这些片段在文库制备的整个过程中保持相互关联。与在dna链断裂位点产生双链断裂的方法(如基于转座子插入的方法)相比,本文公开的方法可避免材料损失并增加目标核酸的克隆覆盖率。
125、第二,与转座子介导的共编码方法相比(例如,如zhang等人,naturebiotechnology,2017年6月,doi10.1038/nbt.3897),切口连接方法避免了转座酶对某些dna序列的偏好造成的偏差。
126、第三,与基于转座子的多步骤共编码方法不同,本文公开的文库制备和共编码过程可作为单步骤、单管制备进行。
127、第四,本文公开的方法所产生的接头片段的大小可通过控制反应中的组分来控制,而不受目标核酸的影响。其他现有的基于转座子的方法产生的靶核酸片段的大小受反应中高分子量基因组dna量的影响,因此往往难以控制。与此相反,在本文公开的方法中,可以通过平衡切口剂和连接酶的量等方法来控制大小。
128、示例性工作流程如图3所示。在步骤1和2中,对双链核酸进行切口处理,以产生交错的单链断裂(220)。在步骤3中,(在没有核苷酸的情况下)通过″间隙酶″(如klenow片段)延长(等同于″加宽″或″打开间隙″)断裂。如图4所示,这些切口和间隙过程会产生单链间隙和重叠的核酸片段(240)(″片段″)。每个片段的一部分与具有互补序列的另一个片段的一部分杂交。
129、在步骤3中,将片段连接到接头上。其中一个接头可以是固定在珠子上的分支连接接头,称为珠联分支连接接头或b-bla。另一个接头可以是溶液中的l-接头。可选地,多余的接头(即未与任何片段连接的接头)可通过核酸酶去除(步骤4)。
130、在步骤5中,在某些情况下,接头片段被加宽以产生包含条形码序列的双链片段。尽管在此作为单独的步骤进行了披露,但裂解和连接可以在单个反应中进行,也可以同时进行。在某些实施方案中,切口和连接反应可持续至少30分钟,例如至少60分钟、至少90分钟或至少120+分钟。在某些实施方案中,双链片段变性形成单链分子。
131、在步骤6中,通过pcr扩增变性的核酸片段,例如使用与片段两端接头序列退火的引物。或者,变性的核酸片段可以循环扩增。
132、本公开还包括该工作流程的各种变化。示例性变化如图5-10所示。
133、iii.方法的示例性实施例
134、可根据各种方案实施切口连接方法。本节提供方法的示例性实施例。具有分子生物学和测序技术技能的从业人员在本公开内容的指导下将认识到个别步骤和试剂的许多变化可纳入以下方案中。
135、方法
136、切口
137、在一种方法中,目标核酸与一种或多种切口剂结合,从而在双链dna中产生交错的单链断裂。在某些实施方案中,切口剂是一种酶(一般称为″切口酶″),例如内切酶,它能裂解多核苷酸链中的磷酸二酯键或从多核苷酸链中去除一个或多个相邻核苷酸。在某些情况下,切口酶是一种非序列特异性内切酶,可在随机位置裂解dna链。切口剂的非限制性实例包括弧菌切口酶(vvn)、虾dsdna特异性内切酶、dnase i、分段酶(mgi)和主酶(masteraseqiagen)。在某些实施方案中,切口剂是一种位点或序列特异性核酸内切酶,如限制性内切酶,可在其识别序列上切口dna。位点特异性核酸酶的非限制性实例包括nt.cvipii(ccd)、nt.bspqi和nt.bbvci,如shuang-yong xu,biomol concepts 2015;6(4):253-267,整个公开内容在此并入作为参考。
138、在某些实施方案中,本文公开的切口也可以是化学切口。化学切口的非限制性实例包括二肽丝氨酰-组氨酸(ser-his)、fe2+/h2o2或cu(ii)复合物/h2o2。
139、因此,切口剂可分为非特异性切口酶、位点特异性切口酶或化学切口剂等类别。在某些实施方案中,该方法使用两种或两种以上的切口剂。在某些实施方案中,该方法使用同一类切口剂中的两种或两种以上切口剂。在某些实施例中,该方法使用不同类别的切口剂。
140、一些参数会影响断裂所分离的核酸片段的长度。通常情况下,切口剂浓度越高,切口处理时间越长,片段长度越短。通过调整这些参数中的一个或多个,可以将片段的长度控制在所需的范围内。在某些实施方案中,核酸片段的平均长度在200到10000个核苷酸之间,例如200-500个核苷酸或400-1000个核苷酸或1000-10000个核苷酸。
141、间隙
142、在某些实施方案中,由切口酶产生的切口会被外切酶延伸(加宽)以形成间隙。这一过程可称为″间隙″,在此过程中使用的外切核酸酶可称为″间隙酶″。具有3′外切酶活性的酶的例子包括dna聚合酶i、klenow片段(在没有核苷酸的情况下)、外切酶iii以及本领域已知的其他酶。具有5′外切酶活性的酶包括bst dna聚合酶、t7外切酶、截短的外切酶viii、λ外切酶、t5外切酶以及本领域已知的其他外切酶。低加工率的外切酶(即以相对较低的速度从多核苷酸末端去除核苷酸的外切酶)最好能打开一个较短的间隙(如2-7个碱基、3-10个碱基或3-20个碱基)并与dna分离,以便进行接头连接。在使用外切酶的情况下,如有必要,可通过在接头的5′端和3′端引入碱基(或修饰碱基)之间的硫代磷酸键来保护dna接头不被外切酶消化。
143、图4展示了使用一种或多种切口剂和一种或多种间隙酶产生重叠核酸片段(240)的过程,这些片段被交错的单链断裂(230)分开。
144、添加接头(连接)
145、如上文所述和图4所示,切口和间隙产生多个片段(240),每个片段具有5′末端和3′末端。在某些实施方案中,″片段″是单链的,尽管如上文和本文其他地方所讨论的,片段可以与互补链杂交,例如形成核酸复合物。第一接头与片段的一个末端(可以是5′末端或3′末端)连接,第二接头(与第一接头不同)与另一个末端连接。结果是多个接头片段具有两种不同的接头序列;反应中产生的所有接头片段具有相同的定义排列(例如,第一接头序列在5′端,第二接头序列在3′端,或者,第二接头序列在5′端,第一接头序列在3′端)。
146、在本公开的一个方面中,第一接头与片段的3′末端连接,第二接头与片段的5′末端连接。在某些实施方案中,第一接头是b-bla,在″3′分支连接″过程中与片段连接。在某些实施方案中,第二接头是″l-接头″。在某些实施方案中,第一接头和第二接头的连接是在与裂解反应和间隙反应相同的反应混合物中进行的。
147、第一接头连接
148、在某些实施方案中,第一接头是bla。bla是本领域已知的,其定义如上。bla包括:(i)双链平末端,包括一条链的5′末端和互补链的3′末端;(ii)单链区域,包括条形码序列。双链平末端提供5′磷酸,可通过3′分支连接到目标核酸片段的3′端。3′分支连接是指将平端接头(供体dna)的5′磷酸酯与双链dna受体的3′羟基端在3′凹股、间隙或切口处共价连接。与传统的dna连接不同,3′分支连接不需要碱基配对。3′分支连接在wang et al.“3′branch ligation:a novel method to ligate non-complementary dna to recessed orinternal 3′oh ends in dna or rna.”dna research 26.1(2019):45-53.;pctpub.no.wo 2019/217452;us pat.pub.us2018/0044668和国际申请wo 2016/037418,uspat.pub.2018/0044667,所有内容均通过引用并入本文。为所有目的,上述各项均并入本文作为参考。
149、使用3′分支连接,理论上可以对捕获的基因组分子的所有子片段进行扩增和测序。因此,3′分支连接具有广泛的分子应用,包括在ngs文库制备过程中将接头连接到dna或rna上等。
150、此外,这一连接步骤还能将样本条形码置于基因组序列附近,以进行多路采样。使用这些接头进行样品条形码编码的好处是,条形码可与基因组dna相邻放置,这样就可以使用相同的引物对条形码和基因组dna进行测序,而无需额外的测序引物来读取条形码。样品条形码可使多个样品的制备物在测序前集中在一起,并通过条形码加以区分。3′支连接接头可按96、384或1536板形式合成,每孔含有多份携带相同条形码的接头,各孔之间的条形码各不相同。在捕获上珠子后,这些接头可用于96、384或1536板形式的连接。
151、3′分支连接可作为一种简单、低成本、无偏差的方法用于标准测序文库的制备,或在有条形码珠(可在条形码接头的3′或5′端连接到珠子)的情况下作为联合条形码文库制备方法进行。这种策略依赖于t4dna连接酶的一个特性,即它可以将双链dna接头连接到dna3′端的切口或间隙,这就是所谓的″3′分支连接″,如wang et al.“3′branch ligation:a novel method to ligate non-complementary dna to recessed or internal 3′ohends in dna or rna.”dna research 26.1(2019):45-53.,并入本文作为参考。由于这种新型连接不需要接头末端的简并单链碱基在间隙中杂交,因此可以在接头结合能力有限的珠子上实现更高效的接头连接。与需要较大间隙(如4-7个碱基)的l-接头连接不同,3′分支连接可以在切口或很小的间隙(1个碱基的间隙)中进行。与5′简并l-接头的连接不同的是,5′简并l-接头的连接可能需要高浓度的5′简并l-接头来补偿连接酶在杂交前不能与l-接头的单链5′-磷酸端结合的事实。
152、为了使珠子上的3′-支链连接效率最高,这些接头可以具有相同碱基的绵延段或简单重复的绵延段,以改善与不完全(如自由松散环)包裹在每个珠子上的目标dna的连接。在将珠子与基因组dna混合之前,可将单链结合蛋白(ssb)与每个接头的单链部分结合。
153、在某些实施方案中,第一接头是b-bla,它包括两条多核苷酸链,在此称为″条形码寡核苷酸″和″杂交寡核苷酸″。条形码寡核苷酸比杂交寡核苷酸长,包含至少一个条形码。条形码寡核苷酸与杂交寡核苷酸杂交,形成部分双链且具有平末端的复合物。
154、在一些实施方案中,条形码寡核苷酸具有5′磷酸,可在分支连接中与3′凹陷片段的3′末端连接,并具有与珠子连接的3′末端;而杂交寡核苷酸不与珠子连接,杂交寡核苷酸为3′阻遏核苷酸(如:双脱氧阻遏核苷酸)。例如双脱氧阻遏核苷酸),阻止磷酸二酯键的形成,从而防止分支接头的自连接。3′分支连接的结果是条形码寡核苷酸连接到片段上。见图11a。
155、在某些实施方案中,杂交寡核苷酸具有5′磷酸,可在分支连接中与3′凹陷片段的3′连接,并具有与珠子连接的3′末端;而条形码寡核苷酸不与珠子连接,条形码寡核苷酸具有阻止形成磷酸二酯键的3′阻断核苷酸。3′分支连接(下文讨论)的结果是杂交寡核苷酸与片段连接见图5和图6。
156、在某些实施方案中,第一接头处于溶液中。在某些实施方案中,部分第一接头固定在珠子上,部分第一接头在溶液中。
157、第二接头连接
158、切口和间隙dna中的片段(相互关联)与第二接头连接。第二接头可以是l-接头、s-bla或任何双链或部分双链接头。
159、在一些实施方案中,第二接头是l-接头。在某些实施方案中,l-接头处于溶液中。l型接头在美国专利no.10,479,991号中有描述,其全部公开内容在此并入作为参考。本方法中使用的l-接头是单链接头,包括杂交区和尾区。l-接头的杂交区包括3′端的简并碱基,如3′端1-10、3-8或4-7个简并核苷酸(ns)。这样,l-接头就能与各种目标序列杂交。当与上述切口dna中的核酸片段接触时,l-接头的杂交区会与目标核酸中的互补序列退火,而尾部区域则保持单链。在允许连接的条件下,l-接头的3′端与核酸片段的5′端连接。例如,参见图5-7。
160、在某些实施方案中,l-接头包括杂交区旁的特定碱基,以提高连接效率和减少假象。例如,如果反应中使用的切口酶优先切割某些碱基或序列,则可将相同的碱基(或互补碱基)设计到l-接头的末端以提高连接效率。在某些实施方案中,可在同一反应中使用具有不同序列(如具有不同数量的简并核苷酸)的两个或多个l-接头。
161、在某些实施例中,第二接头是部分链的接头(图8、9b和10)。在某些实施方案中,第二接头具有双链平末端。在某些实施方案中,在将带切口和间隙的dna中的片段与第一接头连接并通过引物延伸形成双链dna后,可将第二接头连接到与第一接头相反的末端。见图8和图9。在某些实施方案中,第二接头通过平末端连接到片段上。在某些实施方案中,第二接头通过单碱基悬垂连接到片段上,条件是在延伸步骤中使用了留下a尾的聚合酶。
162、在同一反应中将两个接头分别连接到切口或间隙的5′侧和3′侧
163、在某些实施方案中,第一接头(例如b-bla)可以添加到片段的3′末端,第二接头(例如l-接头)可以连接到切口和间隙dna片段的5′末端。而连接是在同样的混合物中进行的,同时也会发生切口和间隙。在某些实施方案中,经过一轮切口连接反应后,可以在额外的第一和/或第二接头存在的情况下,用切口酶和/或间隙酶孵育包有基因组dna的珠子,从而发生第二轮切口连接反应。这种切口连接过程可以重复多轮,例如两轮、三轮或四轮,以提高用两个接头连接的产物的产量。实施例6和7举例说明。
164、可以通过调整l-接头的浓度、温度、循环、ph值、盐浓度、其他添加剂来优化条件,以便在切口中同时连接两个接头,从而增强已连接到基因组片段的分支接头3′端的dna呼吸(dna breathing),使l-接头杂交和连接的单链区域变短。见下文第5节″同时连接的条件″。在一些实施方案中,为了实现更完全的连接,从而获得更多的非重复读数覆盖,除了b-bla外,还可以在溶液中向反应中添加额外的分支连接接头(s-bla)。
165、在某些实施方案中,向反应中加入具有5′外切酶活性的酶,以去除多余的第一接头。这可以在l-接头连接之前进行,也可以与l-接头连接同时进行。由于必须去除多余的接头,因此可以使用较高浓度的l-接头,例如0.01至100μm、0.1至50μm、0.5至30μm、1至20μm,而不会产生大量珠子接头+l-接头连接假象。具有第一接头序列和第二接头序列(如l-接头)的接头片段可以在illumina型系统和其他不需要循环的系统上测序。下文将进一步描述测序的各个方面。
166、在某些情况下,额外的具有3′外切酶活性的酶(如dna聚合酶i、不含核苷酸的klenow片段、外切酶iii或类似物)或具有5′外切酶活性的酶(bstdna聚合酶全长或不含核苷酸的taq聚合酶、t7外切酶、截断的外切酶viii、λ外切酶、t5外切酶或类似物),以增加切口的开放度,为第二接头(例如l-接头)的连接提供更大的空间(例如,t7外切酶viii、λ外切酶、t5外切酶或类似物)。同时具有3′和5′外切酶活性的酶或酶的组合具有优势,即使分支接头在切口中连接,也能为l-接头的连接留出空隙。在使用外切酶的情况下,如有必要,可通过接头5′和3′端碱基和/或修饰碱基之间的硫代磷酸键来实现对dna接头的保护。如上所述,该反应可在聚乙二醇或甜菜碱存在下进行,以提高连接和/或切口酶的活性。
167、此时,如有需要,可按上文所述去除多余的接头。可以使用低浓度的l-接头和其他条件来减少接头与接头之间的连接(如l-接头本身之间的连接或b-bla与l-接头之间的连接),并跳过外切酶去除多余的接头。否则,现在就可以进行pcr,因为子片段的两侧现在都有接头序列。在进行pcr之后,或者如果跳过pcr以实现无pcr版本的流程,那么如上一节所述,下一步就是环化,然后进行滚圆扩增。
168、在一个示例性实施方案中,在单个反应混合物中,非特异性裂解核酸酶、dna连接酶和第一接头、第二接头与双链靶核酸混合,以产生在两个末端都具有接头序列的片段。在优选的实施方案中,第一接头和第二接头中的一个与微米大小的珠子结合,另一个接头在溶液中。
169、在单个反应混合物中加入两个接头的过程可以在溶液中进行,这是一种简单、低成本、无偏差的标准测序文库制备方法。当与附有接头的条码珠子一起使用时,该过程也可用作联合条码文库制备方法。
170、同时切口和连接的条件
171、在一些实施方案中,可以在添加剂(例如聚乙二醇或甜菜碱)的存在下对靶核酸进行切口和间隙,并将一个或多个接头连接到切口和间隙产生的片段上,以提高连接酶的活性、切口剂的活性或两者的活性。在某些实施方案中,连接包括至少将珠子结合的第一接头(如b-bla)连接到核酸片段上。在某些实施方案中,连接包括将溶液中与珠子结合的第一接头和第二接头(如l-接头)都连接到核酸片段上。
172、温度
173、反应可保持在5-65℃范围内的温度,例如5-42℃、10-37℃或5-15℃。在某些实施方案中,反应在室温37℃下进行。在某些实施方案中,当使用热稳定连接酶和切口酶时,反应可保持在高于37℃的温度下进行。在一些实施方案中,反应物在较低温度(5℃-25℃,例如10℃-15℃)和较高温度(例如37℃或更高)之间进行条件循环,循环多次(例如5-100次循环,或20-60次循环,30-55次循环等)。示例见示例1-7。
174、ph
175、在某些实施方案中,反应混合物的ph值保持在5.0至9.0的范围内,例如7.0至9.0,以适应文库制备所需的所有酶功能。切口和连接反应的持续时间可根据所需的核酸片段大小和其他条件(如酶(包括聚合酶、外切酶或两者)浓度、时间、温度、输入dna的量等)而有所不同。
176、时间
177、通常情况下,核酸切口和连接反应的持续时间为5分钟至5小时,例如15-90分钟或30-120分钟。可采用本领域已知的方法终止反应。在某些实施方案中,切口和连接在溶液中进行,反应可通过dna纯化方法(如贝克曼库尔特公司的ampurexp珠)终止。在某些实施方案中,切口和连接是在珠子上进行的,反应可通过用缓冲液(如tris nacl缓冲液)洗涤珠子来终止,以除去切口和连接反应所需的酶和成分。
178、酶
179、本文所述的方法和组合物允许在单个反应混合物中发生切口和连接。在某些实施方案中,选择的条件和酶可使连接发生率高于切口/间隙发生率。这就保证了在随后的连接之前,最初连接的部分切口会被接头连接到其中的大部分切口上,从而最大限度地减少dna的损失。本文公开的方法和组合物允许较高的切口重合率,例如切口重合率为70-100%,例如70-90%、80-90%、80-95%、90-99%)。这里所说的切口重封率是指被打开的切口被连接酶重封的百分比。高切口重封率可通过多种方式实现。在某些实施方案中,使用低活性切口酶进行切口。在某些实施方案中,使用低浓度(例如0000001-10u/ul)的切口酶进行切口。在某些实施方案中,使用具有高连接率的连接酶进行连接。在某些实施方案中,使用高浓度的连接酶进行连接,如1-100u/μl。
180、添加组分的顺序
181、单个反应混合物中各组分的添加顺序可以不同。在某些实施方案中,连接酶在加入酶之前加入或与切口酶同时加入。添加连接酶和将靶核酸加载到珠子上的顺序可以不同。在某些实施方案中,在加入靶核酸(如基因组dna)之前,先将连接酶加入固定有接头的珠子中。在某些实施方案中,先将靶核酸加载到珠上,然后再加入连接酶。
182、在某些实施方案中,最好在加入任何切口、连接酶、之前将靶核酸装载到珠子上,这样靶核酸就会在切口和连接之前与珠子结合。基因组dna包裹在微米大小的顺磁珠上的速度非常快,通常约为1-10分钟。在某些实施方案中,可以采取额外的程序来提高目标核酸与磁珠的结合效率,这对于将长dna(如长度超过200kb的dna)与大磁珠(如直径为3微米或更大的磁珠)结合可能特别有用。在一些实施方案中,目标核酸在含有peg的缓冲液中与珠子结合,peg的浓度相对较高,如3-12%,如5-10%,较高的peg浓度通常会导致较高的结合率。在某些实施方案中,靶核酸在ph值相对较高的缓冲液中与珠子结合,以增强珠子对靶核酸的吸收。在某些实施方案中,ph值大于7.5,例如7.5-9、8.0-9.0或8.0-8.5。高ph值可增加dna的吸附性,尤其是在peg浓度较低的缓冲液中,如5%。在某些实施方案中,缓冲液包含低浓度盐,如10mm mgcl2。本文公开的方法和组合物允许长dna在这些条件下快速(如5-15分钟,大部分dna在1-5或2-10分钟内结合)缠绕珠子,最大限度地减少长dna(如>200kb,或>300kb或>500kb)在与珠子结合前的片段化。例如,长度超过1mb的gdna可与直径约为3um的珠子结合。
183、结合到珠子上的靶核酸仍可进行酶反应,如切口、间隙或接头连接。这样就可以在10-1000个接触点上对与珠子结合的长dna片段(如20-500kb)进行共同编码。这样就可以对吸附在珠子上的dna进行多个连续的酶反应,特别是在如上所述保持dna与珠子结合的条件下。
184、dna可以从珠子中释放出来,为测序做准备。释放dna的方法包括但不限于使用ph值在7-8之间的低盐缓冲液(<200mm),例如约7.5,时间在10分钟至1小时之间,例如约15分钟至约45分钟,约15分钟至约45分钟,或约30分钟。
185、去除多余珠子结合接头的可选步骤
186、可选地,在切口和连接后,使用各种酶去除多余的接头,即未与靶核酸片段连接的接头。在某些实施方案中,与珠子结合的接头是部分双链的,其中每个接头都包括一个相对较短的双链区(如6到20个碱基),并且相对容易变性。也就是说,接头可以在不破坏固定在珠上的双链基因组dna的条件下变性为单链dna。将温度升高到短双链区域的熔点最容易实现这一点。
187、表1显示了可用于去除多余接头的各种酶。
188、表1.可用于去除过量珠结合接头的示例酶
189、
190、然后可以使用外切酶去除变性的单链珠子结合接头。在某些实施方案中,使用外切核酸酶(如recj或exovii)去除过量的与珠子结合的接头,这些接头的3′端部连接到珠子上,这种外切核酸酶可以在5′到3′方向上去除单链dna上的核苷酸。在某些实施方案中,使用可从单链dna中沿3′至5′方向去除核苷酸的外切酶(如exo1、exot)去除多余的珠子结合接头,这些接头的5′末端与珠子连接。
191、或者,不需要变性,多余的、部分双链珠结合的接头可以在没有dntp的情况下,用单链特异性外切酶和对dsdna具有3′到5′外切酶活性的酶的混合物(如exoiii、t4dna聚合酶或phi29dna聚合酶)消化。在本实施方案中,基因组dsdna将通过连接到dna切口或间隙3′端部的接头而免受这些酶的降解。连接产生的核酸片段具有单链末端,而单链末端不是这些dsdna特异性外切酶的底物。
192、在另一种方法中,可以用特定的碱基(如尿嘧啶或肌苷)设计珠结合接头的短双链区,然后用相应的dna糖基化酶(如udg或haag)处理(以产生缺失位点),再用endoiv、endoviii、ape1或任何其他可以去除缺失位点的酶去除这些碱基。使用这种策略,短双链区的熔化温度可以进一步降低,因为在去除这些碱基后,连续双链区的长度会进一步减少。
193、在另一种方法中,如果反应是在溶液中进行的,可以通过dna纯化方法(如ampurexp珠)去除多余的接头。在另一种方法中,当反应在珠子上进行时,多余的接头和与接头连接的产物可通过酶解从珠子上释放出来。在某些实施方案中,珠子结合的接头包括位于珠子近端位置的尿嘧啶或肌苷或两者,可以添加酶来释放这些碱基,从而将接头从珠子中释放出来。在某些实施方案中,接头通过易受化学处理的键与珠子结合,可以添加化学物质来释放接头。例如,接头通过生物素链霉亲和素相互作用与珠子结合,加热或用甲酰胺处理与珠子结合的接头可以破坏这种相互作用。在另一个例子中,接头通过光可裂解连接体与珠子结合,光可用于裂解连接体并将接头从珠子中释放出来。
194、在某些实施方案中,该方法不包括去除多余的珠子结合的接头的步骤;在如上所述的裂解和连接步骤之后,进行引物延伸步骤。在某些实施方案中,引物延伸步骤是在去除多余的珠子连接接头之后进行的。
195、延伸以复制条形码
196、在某些实施方案中,与分支接头连接的核酸片段随后通过dna聚合酶延伸以复制条形码。图8显示了一个说明性的实施例。
197、在某些实施方案中,可以在珠子上或溶液中进行引物延伸步骤,以复制条形码。在某些实施方案中,执行变性步骤(如加热)以产生与接头连接的单链片段,然后使用不具有链置换活性的聚合酶(如pfu、pfucx、taq聚合酶、dna pol 1)延伸与核酸片段连接的链,以复制条形码。图5和图6。在某些实施例中,不进行变性步骤,使用链置换聚合酶(如phi29聚合酶或bst)延长引物。在一个示例中,反应在95℃下变性3分钟,然后使用pfucx在55℃下退火引物3分钟并在72℃下延伸引物10分钟。
198、在某些实施方案中,如果条形码延伸产物在溶液中,可在此步骤中进行另一轮纯化。如果仍与珠子结合,可用tris nacl缓冲液洗涤珠子。
199、如图5和图6所示,在延伸的核酸片段已经包含两个接头(核酸片段的每个末端各一个)的情况下,可将延伸片段从珠子中释放出来,以进行下文所述的进一步处理。在某些实施方案中,延伸产物中只有一个接头,如图8和9a所示,可以将第二个接头连接到核酸片段与第一个接头相反的末端。在某些实施方案中,第二接头通过平末端连接到核酸片段上。在某些实施方案中,如果在延伸步骤中使用了留下a尾的聚合酶,则可以通过单碱基悬垂连接将第二接头连接到核酸片段上。重要的是,为了以无pcr的方式进行连接,接头的3′oh要连接到产物的5′po4上。这是原始的dna链(而不是延伸过程中产生的拷贝)。对于基于pcr的文库预处理策略,通常会在此时进行另一轮dna纯化,然后再进行pcr扩增。
200、控制延伸以分离已连接和未连接的接头
201、受控延伸
202、在另一方面,在将第一接头(例如b-bla)与上述单链断裂分离的核酸片段进行分支连接后,该方法包括在允许控制延伸反应程度的条件下延伸与第一接头序列杂交的引物。这些延伸控制条件包括但不限于选择具有合适聚合速率或其他特性的聚合酶,以及使用各种反应参数,包括(但不限于)反应温度、反应持续时间、引物组成、dna聚合酶、引物和核苷酸浓度、添加剂和缓冲液组成。在某些情况下,延伸可以通过可逆终止子和正常核苷酸的混合来控制。可逆终止子核苷酸量与正常核苷酸量的比例可以调整,以达到延伸的程度;一般来说,可逆终止子核苷酸量与正常核苷酸量的比例越高,延伸越不完全。在某些实施方案中,延伸被控制为只增加约100-150个碱基。
203、在某些实施方案中,引物与第一接头中条形码序列3′的序列杂交,并在延伸控制条件下延伸。在这些条件下,复制连接产物--由第一接头与目标片段连接产生--不完全,导致产生部分双链分子;而复制未连接的b-bla的引物延伸完全,产生双链分子。图12a-12b和图13a-13b展示了使用受控延伸制备接头核酸片段的示例。
204、引物不完全延伸复制连接的第一个接头会留下5′悬垂,可用于3′分支连接。如果使用可逆终止子,在延伸反应结束时,去除可逆终止子的阻断基团以恢复3′oh基团。此时可进行3′分支连接,将第二个接头连接到片段的3′末端,从而产生一个接头片段,该片段的一个末端具有第一个接头序列,另一个末端具有第二个接头序列。在某些实施方案中,可逆终止子可以不同浓度、不同时间点或不同周期添加,以提供核酸片段中大多数核苷酸的重叠覆盖。
205、引物完全延伸复制未连接的第一接头会产生双链分子,可被具有双链dna外切酶活性的酶降解和去除,见表1。
206、去除多余的未连接接头
207、]以下示例方法可用于去除或阻断,或以其他方式尽量减少文库制备中过量未连接接头(即未连接到任何核酸片段的接头)的负面干扰。
208、1.通过纯化珠子去除未连接的接头
209、在某些实施方案中,可以通过ampurexp纯化珠子(beckman coulter,brea,ca)去除溶液中多余的接头。
210、2.用发夹式接头阻断未连接的接头
211、在某些实施方案中,可以使用包括但不限于以下方法的方法降解或阻断多余的接头。图11a和11b中描述的第一种方法使用受控引物延伸,使延伸仅增加约100-150个碱基。这种延伸中使用的聚合酶(如tag聚合酶)不具有3′-5′外切酶活性,它可以产生平末端并在3′末端添加a尾。这将导致多余接头的完全延伸和a尾(即在接头的3′末端添加a)(950),但复制接头片段(940)的延伸将是不完全的。接下来,用发夹式接头进行连接,发夹式接头的t尾与完全延伸的过量接头的a尾互补(950),以阻止这些过量接头的延伸。然而,发夹式接头不能与未完全延伸的接头核酸片段(970)连接。因此,这些片段(970)可以进一步延伸。在某些实施方案中,延伸是在正常核苷酸和可逆终止子的混合物存在下进行的,然后进行反应以去除终止子阻断基团,再用bla(980)进行3′分支连接。现在可以将该产物(990)变性并与珠子分离,保存起来用于测序。珠子可重复用于另一轮使用可逆终止子的引物延伸、去除阻断基团、3′分支连接和变性。这个过程可以重复多次,使用不同浓度的终止子,使基因组片段的dna几乎完全重叠覆盖。
212、3.降解多余的接头
213、]在另一个实施方案中,如图12a和12b所示,使用具有3′-5′外切酶活性的聚合酶(例如pfu、q5、phusion、t7、vent、klenow、t4)进行受控延伸。延伸仅限于约100-150个碱基。同样,结果是连接到基因组片段的接头发生不完全延伸,而多余的接头(即未连接的接头)发生完全延伸。由于聚合酶具有3-5′外切酶活性,结果产生了带有5′磷酸的平端dsdna接头。这是λ外切酶的理想底物,而这些接头片段的不完全延伸产物则不是λ外切酶的理想底物。因此,用λ外切酶处理可以降解所有未连接的多余接头。其余步骤与图11a和11b中描述的步骤基本相同,用于将第二接头连接到基因组片段上。
214、在另一个实施方案中,在控制延伸导致未连接的接头完全延伸和连接产物不完全延伸(图13a)之后,在反应中加入可逆终止子继续控制延伸。一段时间后,从延伸产物中移除终止子阻断基团,并在允许连接的条件下(例如,在连接酶和连接缓冲液存在的情况下)向反应中加入第二个接头。图13b。这将导致多余接头的平端连接和接头片段的3′分支连接。此时,利用链置换聚合酶延长新连接的分支连接接头的一条链(1190),从而进行受控引物延伸。与之前一样,这种延伸受时间、温度和/或核苷酸浓度的控制,只延伸约100-150个碱基。这种延伸会导致第一个接头(如b-bla)的链置换,并释放出一份dsdna接头(1180+1190),可将其与珠子分离并收集起来。与之前的示例一样,可以保存珠子,重复此过程以从每个连接到基因组dna片段的接头生成重叠片段。
215、在某些实施方案中,如上所述(例如,在延伸控制条件下)连接的第一接头延伸后,可以通过平连接或分支连接等方式将第二接头(图10,890)连接到延伸产物的末端。
216、释放
217、两个末端各有一个接头的延伸片段从珠子中释放出来。例如,可以通过降解珠子或裂解接头寡核苷酸与珠子之间的化学连接来实现从珠子中释放。在某些情况下,释放是通过使用endov酶去除捕获寡核苷酸中的肌苷残基,或通过尿嘧啶脱糖苷酶和endoiv/endoviii或其他具有类似功能的酶去除尿嘧啶核苷酸来完成的。在某些情况下,捕获寡核苷酸通过一个或多个二硫键与珠子交联。在这种情况下,可通过将珠子暴露于还原剂(如二硫苏糖醇(dtt)或三(2-羧乙基)膦(tcep))中来实现释放。
218、扩增
219、在某些实施方案中,对上述方法步骤中产生的延伸片段进行扩增。此类扩增方法包括但不限于:多重位移扩增(mda)、聚合酶链反应(pcr)、连接链反应(有时也称为寡核苷酸连接酶扩增ola)、循环探针技术(cpt)、链位移检测(sda)、转录介导扩增(tma)、基于核酸序列的扩增(nasba)、滚动圈扩增(rcr)(用于环化片段)和侵袭裂解技术。扩增可在片段化之后或本文概述的任何步骤之前或之后进行。
220、在图5a和5b中的一个示例中,通过将引物退火至l-接头和分支接头,扩增由目标核酸片段与珠子接头和l-接头连接形成的连接产物。
221、在某些实施方案中,延伸片段可以首先变性为单链核酸分子。然后为每一个单链核酸分子添加一个连接寡聚物,该寡聚物与添加到目标核酸片段两端的接头序列杂交,然后在连接酶(例如t4或taq连接酶)的存在下将单链核酸环化。用于rcr的dna聚合酶可以是任何具有链置换活性的dna聚合酶,例如phi29、bst dna聚合酶、dna聚合酶i的klenow片段和deep-ventr nda聚合酶(neb#mo258)。已知这些dna聚合酶具有不同强度的链置换活性。本领域普通技术人员有能力选择一种或多种合适的dna聚合酶用于本公开的实施例。
222、测序
223、可以使用本领域已知的测序方法对扩增的延伸片段进行测序,包括但不限于基于聚合酶的合成测序(例如,hiseq 2500系统,illumina,san diego,ca)、基于连接的测序(例如,solid 5500,life technologies corporation,carlsbad,ca)、离子半导体测序(例如,ion pgm或ion proton sequencers,life technologies corporation,carlsbad,ca)、零模波导(如pacbio rs测序仪,pacific biosciences,menlo park,ca)、纳米孔测序(如oxford nanopore technologies ltd.,oxford,unitedkingdom)、热测序(如454lifesciences,branford,ct)或其他测序技术。这些测序技术中有些是短读数技术,但也有一些能产生较长的读数,如gs flx+(454life sciences;长达1000bp)、pacbio rs(pacificbiosciences;约1000bp)和纳米孔测序(oxford nanopore technologies ltd.;100kb)。对于单倍型分型,长读数具有优势,需要的计算量更少,尽管它们往往具有更高的错误率,在单倍型分型之前,可能需要根据本文所述的方法识别和纠正这种长读数中的错误。
224、根据一个实施方案,使用组合探针-锚连接(cpal)进行测序,如us 20140051588、u.s.20130124100中所述,为所有目的,这两个文件的全部内容通过引用并入本文。
225、在某些实施方案中,与接头连接的片段或其扩增产物可以变性以产生单链分子。单链分子的两端退火有8-40个碱基的剪接寡核苷酸。这些退火寡核苷酸可使产物的两端有1-10个碱基重叠,类似于限制性酶消化质粒dna后产生的悬垂。然后可以用t4 dna连接酶进行连接,在连接处形成一个带有一小段双链dna的单链圆环。这些圆环现在可用于制造dnbseq测序仪的dna纳米球(dnb)。
226、在某些实施方案中,如上所述,片段的3′末端包含b-bla接头序列,5′末端包含l-接头序列。这些接头片段可在illumina型系统和其他不需要环化的系统上测序。
227、组成
228、1.样品
229、含有目的核酸的样品可以从任何合适的来源获得。例如,样品可以从任何感兴趣的生物体获得或提供。这些生物体包括植物、动物(例如哺乳动物,包括人类和非人类灵长类动物)或病原体,例如细菌和病毒。在某些情况下,样本可以是或可以从这些相关生物群体的细胞、组织或多核苷酸中获得。再比如,样本可以是微生物组或微生物群。可选地,样品是环境样品,如水、空气或土壤样品。
230、来自感兴趣的生物体或感兴趣的生物群的样本可以包括但不限于体液样本(包括但不限于血液、尿液、血清、淋巴、唾液、肛门和阴道分泌物、汗液和精液);细胞;组织;活检、研究样本(例如,核酸扩增反应的产物)、核酸扩增反应的产物,如pcr扩增反应);纯化样品,如纯化基因组dna;rna制剂;以及原始样品(细菌、病毒、基因组dna等)。从生物体中获取目标多核苷酸(如基因组dna)的方法是本领域众所周知的。
231、目标核酸
232、本文所用术语″靶核酸″(或多核苷酸)″或″感兴趣的核酸″是指适合本文所述方法处理和测序的任何核酸(或多核苷酸)。核酸可以是单链或双链,可以包括dna、rna或其他已知核酸。目标核酸可以是任何生物的核酸,包括但不限于病毒、细菌、酵母、植物、鱼类、爬行动物、两栖动物、鸟类和哺乳动物(包括但不限于小鼠、大鼠、狗、猫、山羊、绵羊、牛、马、猪、兔、猴和其他非人灵长类动物以及人类)。目标核酸可从个体或多个个体(即群体)中获得。获取核酸的样本可能包含来自细胞甚至生物体混合物的核酸,如包含人体细胞和细菌细胞的人类唾液样本、包含小鼠细胞和移植人类肿瘤细胞的小鼠异种移植样本等。靶核酸可以是未扩增的,也可以通过本领域已知的任何合适的核酸扩增方法进行扩增。目标核酸可根据本领域已知的方法进行纯化,以去除细胞和亚细胞污染物(脂质、蛋白质、碳水化合物、待测序核酸以外的核酸等),也可以是未纯化的,即至少包括一些细胞和亚细胞污染物,包括但不限于为处理和测序而破坏以释放核酸的完整细胞。目标核酸可以使用本领域已知的方法从任何合适的样本中获得。这些样本包括但不限于生物样本,如组织、分离细胞或细胞培养物、体液(包括但不限于血液、尿液、血清、淋巴、唾液、肛门和阴道分泌物、汗液和精液);以及环境样本,如空气、农业、水和土壤样本等。
233、目标核酸可以是基因组dna(如来自单个个体的dna)、cdna和/或复杂核酸,包括来自多个个体或基因组的核酸。复杂核酸的例子包括微生物组、孕妇血液中的循环胎儿细胞(例如,见kavanagh et al.,j.chromatol.b 878:1905-1911,2010)、癌症患者血液中的循环肿瘤细胞(ctc)。在一个实施方案中,这种复合核酸具有至少一个千兆位(gb)的完整序列(二倍体人类基因组包括约6gb的序列)。
234、在某些情况下,靶核酸或第一复合体是基因组片段。在某些实施方案中,基因组片段长于10kb,例如10-100kb、10-500kb、20-300kb、50-200kb、100-400kb或长于500kb。在某些情况下,靶核酸或第一复合体的长度为5,000至100,000kb。单个混合物中使用的dna(如人类基因组dna)量可小于10ng、小于3ng、小于1ng、小于0.3ng或小于0.1ng dna。在某些实施方案中,单个混合物中使用的dna量可小于单倍体dna量的3,000倍,如小于900倍、小于300倍、小于100倍或小于30倍。在某些实施方案中,单个混合物中使用的dna量可以是单倍体dna量的至少1倍,如至少2倍,或至少10倍。
235、]可以使用常规技术分离目的核酸,例如,如sambrook and russell,molecularcloning:a laboratory manual,cited supra中所披露的那样。在某些情况下,特别是当使用少量核酸时,当仅有少量样品核酸可用,且存在通过非特异性结合(如与容器壁等结合)造成损失的危险时,提供载体dna(如非相关的环状合成双链dna)与样品核酸混合使用是有利的。
236、]根据本公开的某些实施方案,基因组dna或其他复合核酸可通过任何已知方法从单个细胞或少量细胞中获得,无论是否经过纯化。
237、对于本公开的方法来说,长片段是可取的。基因组dna的长片段可通过任何已知方法从细胞中分离出来。例如,peters et al.,nature 487:190-195(2012)中描述了从人类细胞中分离长基因组dna片段的方案。在一个实施方案中,细胞被裂解,完整的细胞核通过温和的离心步骤沉淀下来。然后通过蛋白酶k和rnase消化数小时,释放基因组dna。可通过透析一段时间(即2-16小时)和/或稀释等方法对材料进行处理,以降低剩余细胞废物的浓度。由于这种方法不需要采用许多破坏性过程(如乙醇沉淀、离心和涡旋),基因组核酸基本上保持完整,产生的大部分片段长度超过150千碱基。在某些实施方案中,片段长度约为5至750千碱基。在更多的实施方案中,片段的长度为约150至约600、约200至约500、约250至约400以及约300至约350千碱基。可用于单倍型分析的最小片段约为2-5kb;虽然片段长度会受到起始核酸制备操作产生的剪切力的限制,但并没有最大的理论大小。
238、在其他实施方案中,分离和处理长dna片段的方式可最大限度地减少dna对容器的剪切或吸收,包括例如在琼脂糖凝胶塞或油中分离细胞,或使用特殊涂层的试管和平板。
239、根据另一个实施方案,为了在细胞数量较少(例如,来自微生物检查或循环肿瘤或胎儿细胞的1、2、3、4、5、10、10、15、20、30、40、50或100个细胞)的样品中获得均匀的基因组覆盖率,使用本文公开的方法对从细胞中获得的所有长片段进行条形码编码。
240、条形码
241、根据一个实施方案,使用含有条形码的序列,该序列具有两个、三个或更多的片段,例如,其中一个片段是条形码序列。例如,引入的序列可包括一个或多个已知序列区段和一个或多个作为条形码或标签的简并序列区段。已知序列(b)可包括pcr引物结合位点、转座子末端、限制性内切酶识别序列(如稀有切割器位点,如not i、sac ii、mlu i、bssh ii等)或其他序列。作为标签的简并序列(n)足够长,以提供与待分析的目标核酸片段数目相等或优选大于该数目的不同序列标签群。
242、根据一个实施方案,含条形码序列包括一个任意长度的已知序列区域。根据另一个实施方案,含条形码序列包括选定长度的已知序列两个区域,其侧翼是选定长度的简并序列区域,即bnnnbn,其中n可以具有足以标记目标核酸长片段的任何长度,包括但不限于n=10、11、12、13、14、15、16、17、18、19或20,而b可以具有容纳所需序列如转座子末端、引物结合位点等的任何长度。例如,这样的实施方案可以是b20n15b20。
243、在一个实施方案中,用于标记长片段的条形码采用两段或三段设计。这种设计允许通过将不同的条形码片段连接在一起形成完整的条形码片段,或在寡核苷酸合成中使用片段作为试剂,从而产生组合条形码片段,从而提供更多可能的条形码。这种组合设计提供了更多可能的条形码,同时减少了需要生成的全尺寸条形码的数量。在进一步的实施方案中,使用8-12个碱基对(或更长)的条形码可实现每个长片段的唯一识别。
244、在一个实施例中,使用了两个不同的条形码段。a段和b段很容易修改,各自包含不同的半条形码序列,从而产生数千种组合。在另一个实施例中,条形码序列包含在同一个接头上。这可以通过将b型接头分成两部分来实现,每一部分都包含一个半条形码序列,中间用一个用于连接的共同重叠序列隔开。两个标签部分各有4-6个碱基。一个8碱基(2x4碱基)的标签组能够唯一标记65,000个序列。2x5个碱基和2x6个碱基的标签都可以使用简并碱基(即″通配符″),以达到最佳解码效率。
245、在更多的实施方案中,每个序列的唯一识别是通过8-12个碱基对的纠错条形码来实现的。条形码的长度可为5-20个信息碱基,通常为8-16个信息碱基。
246、umi
247、在不同的实施方案中,唯一分子标识符(umi)用于区分单个dna分子。例如,umi用于区分固定在第一珠上的捕获寡核苷酸。生成的接头集合每个都有一个umi,这些接头连接到待测序的片段或其他源dna分子上,每个测序分子都有一个umi,有助于将其与所有其他片段区分开来。在这种情况下,可以使用大量不同的umi(例如,数千到数百万个)来唯一识别样本中的dna片段。
248、umi的长度足以确保每个源dna分子的唯一性。在某些实施方案中,唯一分子标识符的长度约为3-12个核苷酸,或3-5个核苷酸。在某些情况下,每个唯一分子标识符的长度约为3-12个核苷酸,或3-5个核苷酸。因此,唯一分子标识符的长度可以是3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18或更多核苷酸。
249、条形编码珠
250、珠子由固定在其上的b-bla中的条形码寡核苷酸编码。每个珠子包含多个b-bla,因此也包含多个条形码寡核苷酸。每个条形码寡核苷酸包括至少一个条形码。同一珠子上的条形码寡核苷酸具有相同的条形码序列,而不同珠子上的条形码寡核苷酸具有不同的条形码序列。因此,每个珠子都携带有许多份独特的条形码序列,可使用上述方法将其转移到靶核酸片段上。
251、所用珠子的直径可在1-20微米范围内,也可为2-8微米、3-6微米或1-3微米,例如约2.8微米。例如,珠子上条形码寡核苷酸的间距可以是至少1nm、至少2nm、至少3nm、至少4nm、至少5nm、至少6nm或至少7nm。在一些实施方案中,间距小于10nm(例如5-10nm)、小于15nm、小于20nm、小于30nm、小于40nm或小于50nm。在某些实施例中,每种混合物使用的不同条形码的数量可以大于100万、大于1000万、大于1000万、大于1000万、大于1000万、大于1000万、大于1000万或大于1000万。如下文所述,本公开内容中可以使用大量条形码,例如使用本文所述的方法。在一些实施例中,每种混合物使用的不同条形码的数量可以>100万、>1000万、>3000万、>1亿、>3亿或>1b,并且它们是从至少10倍以上的多样性池中采样的(例如,从>1000万、>0.1b、0.3b、>0.5b、>1b、>3b、>10b珠子上的不同条形码中采样)。在一些实施例中,每个珠子上的条形码数量在100k到10m之间,例如,在200k到1m之间,在300k到800k之间,或约400k之间。
252、在一些实施方案中,条形码区域的长度约为3-15个核苷酸,例如5-12、8-12或10个核苷酸。在某些情况下,条形码区的每个条形码长度约为3-12个核苷酸,或3-5个核苷酸。因此,条形码(无论是样品条形码、细胞条形码还是其他条形码)的长度可以是3、4、5、6、7、8、9、10、11、12、13、14、15或更多核苷酸。在一个例子中,每个条形码区域包括三个条形码,每个条形码由10个碱基组成,三个条形码之间由6个碱基的共同序列隔开。
253、条形码珠被转移到目标核酸序列上。在某些实施方案中,通过将接头寡核苷酸的3′末端与通过所公开的切口和间隙形成的核酸片段连接,以固定的时间间隔进行转移。
254、在某些实施方案中,条形码珠是通过使用三组双链条形码dna分子的分裂和汇集连接策略构建的。在某些实施方案中,每组双链条形码dna分子由10个碱基对组成,三组分子的核酸序列不同。pct pub.no.wo2019/217452中描述了生产条形码珠子的示例性方法,其公开内容在此全文并入作为参考。并入本文作为参考的wo2019/217452的图12和13也说明了拆分和汇集法的方法。在一种方法中,将包含pcr引物退火位点的通用接头序列连接到带有5′双生物素连接头的dynabeadstm m-280 streptavidin(thermofisher,waltham,ma)磁珠上。integrated dna technologies(coralville,ia)构建了三组1536个条形码寡聚体,其中包含序列重叠区域。连接在384孔板中的15μl反应液中进行,反应液含有50mmtris-hcl(ph7.5)、10mm mgcl2、1mm atp、2.5%peg-8000、571unit t4连接酶、580pmol条形码寡聚体和6500万m-280珠子。连接反应在室温下旋转孵育1小时。在两次连接之间,通过离心将珠子集中到一个容器中,用磁铁收集到容器的一侧,然后用高盐洗涤缓冲液(50mmtris-hcl(ph7.5)、500mm nacl、0.1mm edta和0.05%tween20)洗涤一次,用低盐洗涤缓冲液(50mm tris-hcl(ph7.5)、150mm nacl和0.05%tween20)洗涤两次。将珠子重悬于1x连接缓冲液中,并分布于384孔板中,重复连接步骤。
255、在一个方面,本公开提供了一种组合物,该组合物包含附有包含克隆条形码的接头寡核苷酸的珠子,其中组合物包含30亿个以上不同的条形码,条形码是结构为5′-cs1-bc1-cs2-bc2-cs3-bc3-cs4的三方条形码。在某些实施例中,cs1和cs4比cs2和cs3长。在某些实施方案中,cs2和cs3的长度为4-20个碱基,cs1和cs4的长度为5或10至40个碱基,例如20-30个碱基,而bc序列的长度为4-20个碱基(例如10个碱基)。在某些实施方案中,cs4与剪接寡核苷酸互补。在某些实施方案中,组合物包含桥寡核苷酸。在某些实施方案中,组合物包含桥寡核苷酸、包含上文讨论的三方条形码的珠子和包含与桥寡核苷酸互补区域的杂交序列的基因组dna。
256、克隆条形码的另一种来源,如与多个标签拷贝相关联的珠子或其他支持物,可通过乳液pcr或cpg(可控孔玻璃)或化学合成制备,其他颗粒带有由以下方法制备的适应条形码拷贝。含标签的dna序列群可以通过已知的方法在油包水(w/o)乳液中的珠子上进行pcr扩增。例如,见tawfik and griffiths nature biotechnology 16:652-656(1998);dressman et al.,proc.natl.acad.sci.usa 100:8817-8820,2003;以及shendure etal.,science 309:1728-1732(2005)。这样,每个珠子上的每单个含标签序列都有许多拷贝。
257、制作克隆条形码源的另一种方法是在微珠或cpg上以″混合和分割″组合工艺合成寡核苷酸。使用这种方法可以制造出一组微珠,每组珠子都有一个条形码的群体拷贝。例如,要制作所有的b20n15b20,平均每100个珠子上有约1000多个拷贝,其中每个珠子上有约10亿个拷贝,可以从约1,000亿个珠子开始,在所有珠子上合成b20共用序列(接头),然后将它们分成1024个合成柱,在每个柱子上制作不同的5-mer,然后将它们混合,再将它们分成1024个柱,制作更多的5-mer,然后再重复一次,完成n15,然后将它们混合,在一个大柱中合成最后一个b20作为第二个接头。这样,在3050次合成中,就能做出与用~1000亿个珠子(112个珠子)进行的大型仿真pcr反应相同的″克隆式″条形码集,因为10个珠子中只有1个有起始模板(其他9个没有),以避免每个珠子有两个不同条形码的模板。
258、pct pub.no.wo2019/217452中描述了条形码序列组装的示例过程,其公开内容在此并入作为参考。
259、固定
260、多核苷酸可以通过多种技术固定在基底(例如珠子)上,包括共价和非共价附着。多核苷酸可以通过多种技术固定到基底上。在某些实施方案中,多核苷酸与基底(如珠子)连接,即多核苷酸的一个末端直接与基底接触或连接。例如,表面可能具有活性官能团,与多核苷酸分子上的互补官能团发生反应,形成共价连接。长dna分子,例如几个核苷酸或更大的dna分子,也可以有效地附着在疏水表面上,例如具有低浓度各种活性官能团(如-oh基团)的洁净玻璃表面。在另一个实施方案中,多核苷酸分子可通过与表面的非特异性相互作用或通过氢键、范德华力等非共价相互作用吸附到表面上。
261、在一些实施方案中,多核苷酸通过与表面上的捕获寡核苷酸杂交,并与捕获寡核苷酸的组分形成复合物,例如双链双工物或部分双链双工物,从而固定到表面上。
262、反应混合物
263、本文提供的反应混合物包含一种或多种切口剂、一种或多种连接酶、多个珠子、多个被交错单链断裂分隔的重叠核酸片段。每个珠子包括至少一个固定在其上的分支连接接头。每个分支连接接头包括一个杂交寡核苷酸和一个条形码寡核苷酸。条形码寡核苷酸包括条形码并与珠子连接,而杂交寡核苷酸不与珠子连接。多个珠子中的每一个都包含唯一的条形码序列,即同一珠子上的分支连接接头具有相同的条形码序列,而不同珠子上的分支连接接头具有不同的条形码序列。
264、条形码寡核苷酸与杂交寡核苷酸杂交,形成部分双链核酸分子,包括单链区和双链区。双链区包括具有5′末端和3′末端的双链平末端,双链平末端的5′末端与核酸片段的3′末端连接。
265、本公开中引用的每份出版物和专利文件都通过引用并入本文,如同每份此类出版物或文件都具体和单独地指明通过引用并入本文。对出版物和专利文件的引用并不表示任何此类文件是相关的现有技术,也不构成对其内容或日期的承认。
- 还没有人留言评论。精彩留言会获得点赞!