一种5,6-二羟基吲哚啉和5,6-二羟基吲哚的选择性制备方法与流程
本发明涉及一种5,6-二羟基吲哚啉和5,6-二羟基吲哚的选择性制备方法,属于药物有机合成。
背景技术:
1、目前市面上的染发产品种类繁杂,其主要的有效染黑成分为对苯二胺及其衍生物,这类物质对人体具有致癌性、致畸性、致过敏、遗传毒性等不良反应,严重影响人们的身体健康。因此,迫切需要安全有效且使用便利的新型染发剂。
2、5,6-二羟基吲哚啉是生物体毛囊当中的黑色素细胞合成黑色素的一种重要的中间体。生理条件下,它在生物体内会进一步氧化、聚合生成复杂的具有颜色的高分子物质黑色素;在体外弱碱性环境下或是有氧环境中,也会发生同样的变化。现有技术中已经报道了以5,6-二羟基吲哚啉氢溴酸盐作为人发染色剂当中的染发成分,对角蛋白纤维具有很好的染色作用。此外,中国专利文献cn105380892a公开了一种复合单剂型染发剂,5,6-二羟基吲哚啉是其复合单剂型染发剂中黑色素前驱体中的几种有效成分之一。因此,5,6-二羟基吲哚啉作为新一代安全绿色的染发剂的有效成分,具有很高的商业价值和工业应用价值。
3、近年来,随着精细化学品合成的发展,5,6-二羟基吲哚啉不仅可以作为一些生物碱、氨基酸等制备过程中的中间体,其在其他方面的应用也不断出新,例如,5,6-二羟基吲哚啉可以作为很多药物中有效成分的合成原料或重要中间体;还可以作为除菌剂、抗氧化剂和防腐剂等一些添加剂中的有效成分。
4、但是,5,6-二羟基吲哚啉性质不稳定,不仅对ph、温度以及氧气十分敏感,而且在合成工艺上存在一定问题。中国专利文献cn107540596a公开了一种化合物5,6-二羟基吲哚啉及其氢卤酸盐的制备方法,具体是以3,4-二烷氧基苯乙胺为原料合成5,6-二羟基吲哚啉及其氢卤酸盐方法,但是由于5,6-二羟基吲哚啉性质极不稳定,其最终产物制备成了氢卤酸盐的形式。此外,文献journal of medicinalchemistry,1995,vol.38(6),917-922中报道了一种以3,4-二甲氧基苯乙腈为原料的合成方法,经过硝化反应、氢化还原和酸溶液高温去保护基等化学反应得到最终产物5,6-二羟基吲哚啉。但是该方法存在原料成本高,硝化反应后处理繁琐,氢化还原后的副产物多且还原试剂价格昂贵等问题,在实际的操作和生产中比较困难。
5、5,6-二羟基吲哚也是一种天然的黑色素原的重要中间体,它对人体皮肤刺激小,安全性高,作用机制明确,正逐步取代苯胺类化合物作为新型染发剂的最佳选择。因此,5,6-二羟基吲哚同样作为新一代安全绿色染发剂的有效成分,也具有很高的商业价值和工业应用价值。
6、基于5,6-二羟基吲哚在染发剂、抗氧化剂应用方面取得的良好作用,上世纪90年代初期欧莱雅、宝洁、花王等世界大型化妆品公司就已经开始着手于研制5,6-二羟基吲哚。上世纪90年代中期,国内一部分化妆品公司也开始陆续委托科研机构进行有关5,6-二羟基吲哚的研究工作,但是由于5,6-二羟基吲哚对氧气、过渡金属、酸碱试剂等较为敏感,性质很不稳定易变质,并且已有的生产方法由于收率不高以及操作条件苛刻等原因,一直难以制备出高纯度的5,6-二羟基吲哚。
7、文献inorg.chem.2006,45,3657-3664报道了以左旋多巴为原料一步合成5,6-二羟基吲哚的方法,该合成方法用水量巨大,产生大量废水,处理成本高。文献《精细石油化工》,2007,24(2):53-55报道了以3,4-二甲氧基苄腈为原料通过去甲基、羟基保护、硝化、还原、环化等多步反应得到5,6-二羟基吲哚,虽然这种方法产率较高,可达到80%以上,但制备环境条件苛刻,工业放大存在一定困难。文献hair dye composition.2006,02,10以多巴胺类化合物为起始原料,通过使侧链与苯环上邻位取代基的环化得到5,6-二羟基吲哚。这类方法虽然反应步骤少,但所用原料价格较高,羟基裸露后性质极不稳定,产率仅为10%,经济性较差且所得产物纯度低,不适合运用到工艺大生产之中。因此急需研发一种低成本的、工业化的5,6-二羟基吲哚啉和5,6-二羟基吲哚制备方法。
技术实现思路
1、针对现有技术的不足,本发明提供一种5,6-二羟基吲哚啉和5,6-二羟基吲哚的选择性制备方法。
2、本发明的技术方案如下:
3、一种5,6-二羟基吲哚啉和5,6-二羟基吲哚的选择性制备方法,包括步骤如下:
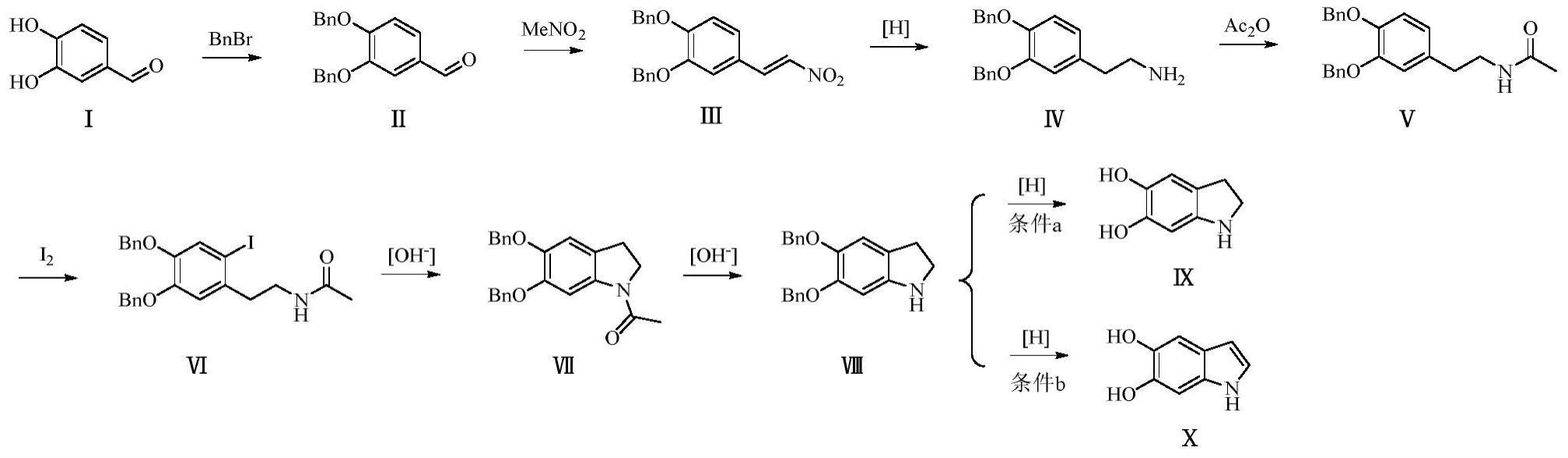
4、(1)将3,4-二羟基苯甲醛和溴化苄溶于有机溶剂中,在ph为8~11、30~80℃下反应8~10h,然后将反应体系冷却至20~30℃后稀释,经萃取、干燥和减压蒸除溶剂后,得到化合物ⅱ;
5、(2)将乙酸、硝基甲烷和乙酸铵混合均匀,在20~30℃、搅拌条件下加入化合物ⅱ混合均匀后,升温至80~130℃,反应8~11h,然后将反应体系冷却至20~30℃后倒入去离子水中析出固体,经过滤、洗涤、打浆和真空干燥后,得到化合物ⅲ;
6、(3)在氮气保护和冰浴搅拌条件下,将还原剂溶于有机溶剂中,配成溶液a;
7、(4)在氮气保护和搅拌条件下,将化合物ⅲ溶于有机溶剂中,配成溶液b;
8、(5)在氮气保护和冰浴搅拌条件下,将溶液b缓慢滴入溶液a中,在10~60℃下反应3~4h,然后将反应体系冷却至20~30℃,在冰浴搅拌条件下,继续滴加碱性溶液,调节ph至8~11,过滤,滤液经干燥和减压蒸除溶剂后,得到化合物ⅳ;
9、(6)将化合物ⅳ溶于有机溶剂中,于冰浴搅拌条件下滴加酰化试剂和缚酸剂,在20~30℃反应3~4h,然后将反应液稀释后进行萃取、干燥和减压蒸除溶剂,得到化合物v;
10、(7)将碘溶于有机溶剂中,配成溶液c;将化合物v溶于有机溶剂中,在20~30℃、搅拌条件下向化合物v的有机溶液中滴加溶液c,然后在40~50℃下反应8~10h,接着将反应体系冷却至20~30℃,用碱性溶液调节ph至8~9,经洗涤、萃取、干燥和减压蒸除溶剂后,得到化合物ⅵ;
11、(8)将化合物ⅵ溶于有机溶剂中,加入催化剂,在氮气保护、110~160℃下反应3~4h,然后将反应体系冷却至20~30℃后稀释,经萃取、干燥和减压蒸除溶剂后,得到化合物vii;
12、(9)将化合物vii溶于醇溶液中,在氮气保护、ph为8~11、50~100℃下反应4~5h,然后将反应体系冷却至20~30℃后析出固体,经过滤、洗涤、结晶和真空干燥后,得到化合物ⅷ;
13、(10)将化合物ⅷ溶于有机溶剂中,加入还原剂,在h2保护、20~30℃下反应2~3h,将反应液在无氧环境中过滤,滤液减压蒸除溶剂,得到5,6-二羟基吲哚啉;
14、(11)将化合物ⅷ溶于有机溶剂中,加入还原剂,在h2保护、20~30℃下反应2~3h,将反应液在无氧环境中过滤,滤液减压蒸除溶剂,得到5,6-二羟基吲哚。
15、根据本发明优选的,步骤(1)中,所述3,4-二羟基苯甲醛和溴化苄的质量体积比为8:(20~21),单位:g/ml;所述有机溶剂为二氯甲烷、三氯甲烷、乙醚、乙酸乙酯、甲醇、乙醇、n,n-二甲基甲酰胺中的一种或几种;所述有机溶剂和溴化苄的体积比为30:(20~21)。
16、进一步优选的,所述有机溶剂为n,n-二甲基甲酰胺。
17、根据本发明优选的,步骤(1)中,通过氢氧化钾、氢氧化钠、碳酸钠、碳酸钾或二甲基吡啶调节ph为8~11;反应温度为40~60℃。
18、进一步优选的,通过碳酸钾调节ph为8~11,反应温度为50℃。因碳酸钾廉价易得,可以进一步降低成本,反应温度为50℃时化合物ⅱ产率最高。
19、根据本发明优选的,步骤(1)中,所述稀释、萃取、干燥和减压蒸除溶剂具体为:
20、将反应体系用二氯甲烷进行稀释,然后用去离子水、饱和食盐水萃取,再用无水硫酸钠干燥,减压蒸除溶剂得化合物ⅱ;所述二氯甲烷和溴化苄的体积比为50:(20~21)。
21、进一步优选的,所述减压蒸除溶剂后,将向化合物ⅱ中加入乙醇打浆15~30min,过滤,用冷乙醇洗涤滤饼,60℃真空干燥,得到纯化后的化合物ⅱ。通过乙醇打浆可以进一步纯化化合物ⅱ。
22、根据本发明优选的,步骤(2)中,所述乙酸、硝基甲烷和乙酸铵的体积质量比为(60~62):(10~13):(5~6),单位:ml/ml/g;所述和化合物ⅱ和乙酸铵的质量比为(14~15):(5~6);所述乙酸和去离子水的体积比为(30~31):(25~40)。
23、根据本发明优选的,步骤(2)中,所述过滤、洗涤和真空干燥具体为:
24、将析出固体过滤后,用乙醇清洗滤饼,然后向滤饼中加入乙酸乙酯打浆15~30min,再次过滤,滤饼用滤液洗涤,60℃真空干燥,得到化合物ⅲ。
25、根据本发明优选的,步骤(3)中,所述还原剂为硼氢化钠、四氢铝锂、醋酸钯、氢氧化钯、雷尼镍中的一种或几种;所述有机溶剂为醚类溶剂;所述溶液a的浓度为30~40g/l。
26、进一步优选的,所述还原剂为四氢铝锂,所述有机溶剂为乙醚。四氢铝锂做还原剂产率最高,同时四氢铝锂在乙醚中更稳定。
27、根据本发明优选的,步骤(4)中,所述有机溶剂为四氢呋喃;所述溶液a的浓度为120~130g/l。
28、根据本发明优选的,步骤(5)中,所述溶液b和溶液a的体积比为(22~23):(34~35);所述反应温度为40℃;所述碱性溶液为10%的naoh溶液,调节ph为10。
29、根据本发明优选的,步骤(6)中,所述有机溶剂为二氯甲烷、三氯甲烷、乙醚、乙酸乙酯、甲醇、乙醇、n,n-二甲基甲酰胺中的一种或几种;所述酰化试剂为乙酰氯、乙酸、乙酸酐中的一种或几种;所述缚酸剂为吡啶、二乙胺、三乙胺中的一种或几种;
30、所述化合物ⅳ和有机溶剂的质量体积比为1:6,单位:g/ml;所述有机溶剂、酰化试剂和缚酸剂的体积比为72:(6~7):(7~8)。
31、进一步优选的,所述有机溶剂为n,n-二甲基甲酰胺;所述酰化试剂为乙酸酐;所述缚酸剂为三乙胺。
32、根据本发明优选的,步骤(7)中,所述有机溶剂为二氯甲烷、三氯甲烷、乙醚、乙酸乙酯、甲醇、乙醇、n,n-二甲基甲酰胺中的一种或几种;所述溶液c的浓度为1~1.2g/ml;所述化合物v和有机溶剂的质量体积比为(13~14):(117~118),单位:g/ml;所述化合物v和溶液c的质量体积比为(13~14):(26~27),单位:g/ml;所述碱性溶液为0.4m的naoh溶液。
33、进一步优选的,所述有机溶剂为n,n-二甲基甲酰胺。
34、根据本发明优选的,步骤(8)中,所述有机溶剂为甲醇、乙醇、异丙醇、乙醚、二氯甲烷、三氯甲烷、二甲基亚砜中的一种或几种;所述催化剂为碘化亚铜和碳酸钾的混合物;所述化合物ⅵ和有机溶剂的质量体积比为(16~17):220,单位:g/ml;所述化合物ⅵ、碘化亚铜和碳酸钾的质量比为(16~17):(1~1.5):(13~14)。
35、进一步优选的,所述有机溶剂为二甲基亚砜。
36、根据本发明优选的,步骤(9)中,所述醇溶液为无水甲醇、无水乙醇、异丙醇、正丙醇、乙二醇中的一种或几种;所述化合物vii和醇溶液的质量体积比为(1.2~1.3):(21~22),单位:g/ml;所述ph为8~11是通过氢氧化钠或氢氧化钾的饱和溶液调节;所述反应温度为90℃。
37、进一步优选的,所述醇溶液为无水乙醇。
38、根据本发明优选的,步骤(10)中,所述有机溶剂为四氢呋喃、无水甲醇、无水乙醇、异丙醇、乙酸乙酯、1,4-二氧六环中的一种或几种;所述还原剂为硼氢化钠、四氢铝锂、醋酸钯、氢氧化钯炭、雷尼镍、钯碳中的一种或几种;所述化合物ⅷ、有机溶剂和还原剂的质量体积比为(3~6):200:(0.2~0.8),单位:g/ml/g。
39、进一步优选的,所述有机溶剂为无水甲醇,还原剂为氢氧化钯炭。
40、根据本发明优选的,步骤(11)中,所述有机溶剂为四氢呋喃、无水甲醇、无水乙醇、异丙醇、乙酸乙酯、1,4-二氧六环中的一种或几种;所述还原剂为硼氢化钠、四氢铝锂、醋酸钯、氢氧化钯炭、雷尼镍、钯碳中的一种或几种;所述化合物ⅷ、有机溶剂和还原剂的质量体积比为(80~120):1~2:(8~12),单位:mg/ml/mg。
41、进一步优选的,所述有机溶剂为乙酸乙酯,还原剂为氢氧化钯炭。
42、本发明未详尽之处,均可采用现有技术。
43、本发明的有益效果在于:
44、1、本发明以廉价的3,4-二羟基苯甲醛作为起始原料,采用常见的催化剂,依次通过苄基保护、硝化、还原、酰胺化、卤素取代、关环、水解以及还原反应得到5,6-二苄氧基吲哚啉,然后再以5,6-二苄氧基吲哚啉为反应物,通过添加不同的有机溶剂和还原剂能够选择性的制备得到5,6-二羟基吲哚啉和5,6-二羟基吲哚,有效克服了现有技术中只能单路径制备5,6-二羟基吲哚的问题。并且本发明制备5,6-二羟基吲哚啉和5,6-二羟基吲哚的综合产率分别达到了23%和28%,相较于现有技术降低了成本,提高了产率。
45、2、本发明提供了一种新的制备5,6-二羟基吲哚啉和5,6-二羟基吲哚的反应路线和制备方法,反应试剂更加环保安全,原料廉价易得,反应产率高,副反应少,适于工业化生产。
- 还没有人留言评论。精彩留言会获得点赞!