一种FGFR2抑制剂Lirafugratinib(RLY-4008)的制备方法与流程
本发明属于药物化学合成领域,涉及一种fgfr2抑制剂lirafugratinib(rly-4008)的新的制备方法。
背景技术:
1、胆管癌(cca)作为一种罕见的恶性肿瘤,预后较差。恶性程度和公认的癌王胰腺癌不相上下。这类癌症及时接受手术治疗,3年复发率也达到85%,患者5年生存率仅有约5%;而无法手术切除的患者5年生存率一度低至0。fgfr2是一种受体酪氨酸激酶,fgfr家族的四个成员之一,是一组密切相关的蛋白质,具有高度相似的蛋白质序列和特性,在许多癌症中经常发生突变,包括肝内胆管癌(10%-16%)、子宫内膜癌(7.5%-11%)、胃或胃食管交界处腺癌(3.7%-7.9%)等。fgfr2的致癌激活可以通过基因扩增、激活突变或染色体重排,胆管癌的fgfr2融合/重排发生率为10-15%的。之前的非选择性fgfr抑制剂通过实现20-40%的客观缓解率(orr)和5-9个月的缓解持续时间(dor)验证了fgfr作为cca的治疗靶点。但是泛fgfr抑制剂(infigratinib,pemigatinib,futibatinib)的二线治疗患者常常出现高磷血症和腹泻等不良反应,同时还会发生fgfr2耐药突变,需要多方面调整剂量,难已达到药物最佳剂量,对于fgfr2的选择性抑制有望提供优越的目标覆盖率,从而大幅提高药物疗效。
2、尽管在基于结构的药物设计方面进行了大量研究,但fgfr2的选择性靶向仍然存在未知的类似于fgfr1、fgfr3和fgfr4的激酶结构域,rly-4008是第一个高选择性、强效的fgfr2抑制剂,基于独特的构象动力学选择性地针对驱动程序改变和fgfr抗性突变,具有更强的敏感性和体内活性。rly-4008的i/ii期临床试验结果显示治疗胆管癌的有效率达到了88%,且没有显示出高磷血症(fgfr1)和腹泻(fgfr4)的非目标毒性的限制。这些初步数据表明,rly-4008具有可控的安全性,并能推动多种fgfr2突变和肿瘤类型的肿瘤消退。
技术实现思路
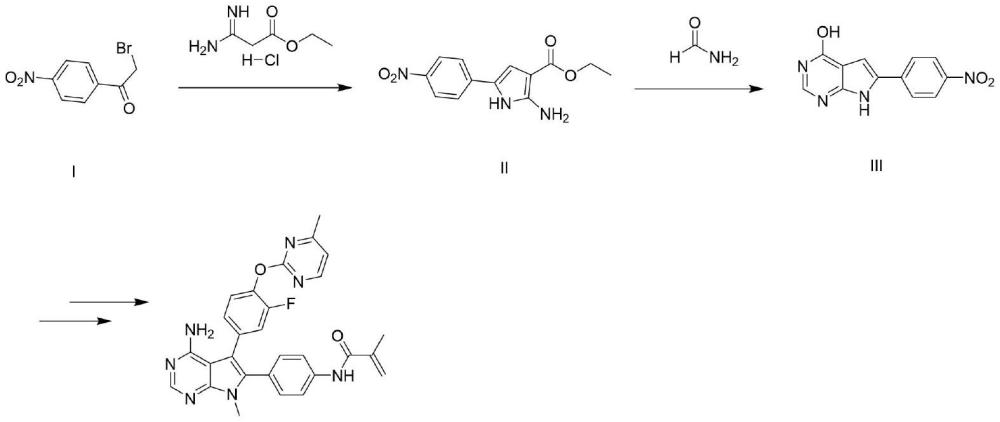
1、鉴于上述情况,fgfr2抑制剂lirafugratinib(rly-4008)的制备非常重要。本发明人通过实验研究解决了该化合物的技术问题,其反应路线如下:
2、
3、具体实验方式
4、下面通过实施例对本发明作进一步描述说明,但并不因此而限制本发明的内容。
5、实例1:
6、
7、步骤a
8、将化合物3-脒基丙酸乙酯盐酸盐(8.3g,50.0mmol)和naoet(5.1g,75.0mmol)溶于无水乙醇中,并在氩气条件下搅拌20min,将混合物加热至60℃,并在5分钟内分批加入2-溴代对硝基苯乙酮(6.1g,25.0mmol),反应1.5h后,混合物冷却至20℃,减压蒸发溶剂。残留物用蒸馏水(20ml)稀释,并用乙酸乙酯(3×80ml)萃取。有机层用水(3×20ml)和盐水(3×20ml)洗涤。合并的水溶液再用etoac(2×20ml)萃取。有机相mgso4干燥,减压蒸干。残余物通过硅胶柱层析纯化得到2-氨基-5-(4-硝基苯基)-1h-吡咯-3-羧酸乙酯(4.14g,产率60%)。lc-ms(esi):m/z=275.3[m+h]+.
9、步骤b
10、在无水dmf(28ml)溶液中加入甲酸(11.3ml)、2-氨基-5-(4-硝基苯基)-1h-吡咯-3-羧酸乙酯(4.60g,16.57mmol)和过量甲酰胺(75ml),升温至120℃反应20小时。然后加入2-丙醇(12ml),混合物冷却至20℃。过滤,用2-丙醇(10ml)和正己烷(2×15ml)洗涤,减压干燥,得到化合物4-羟基-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(2.52g,产率60%)。lc-ms(esi):m/z=256.2[m+h]+
11、步骤c
12、将4-羟基-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(1.9g,7.43mmol)和pocl3(13.3ml)混合,在90℃下反应3小时。用冰盐浴冷却,然后加入水(60ml)。用naoh(8m,80ml)将ph值调至12。过滤,用水和正戊烷洗涤,干燥后得到化合物4-氯-6-(4-硝基苯基)-7h-吡咯并[2,3-d]嘧啶(1.93g,产率92%)。lc-ms(esi):m/z=274.7[m+h]+
13、步骤d
14、将4-氯-6-(4-硝基苯基)-7h-吡咯并[2,3-d]嘧啶(179mg,0.653mmol)和碳酸铯(319mg,0.980mmol)溶于无水dmf(2ml)中,在30min内加入碘甲烷(0.65ml,1.31mmol,2m在叔丁基甲基醚中),溶液在室温下搅拌反应90min。用h2o(50ml)淬灭反应,并用etoac(2×30ml)萃取。合并的有机相用饱和nahco3(15ml)和饱和nacl(20ml)洗涤,用无水硫酸钠干燥,过滤并浓缩。残余物通过硅胶柱层析纯化,得到化合物4-氯-7-甲基-6-(4-硝基苯基)-7h-吡咯并[2,3-d]嘧啶(169mg,产率90%)。lc-ms(esi):m/z=288.7[m+h]+步骤e
15、将化合物4-氯-7-甲基-6-(4-硝基苯基)-7h-吡咯并[2,3-d]嘧啶(531g,1.84mol)悬浮于在氨水(30%h2o中,3.63l)中,在压力容器中搅拌下120℃反应18h,冷却到20℃,过滤,用h2o(1.80l)和甲醇(900ml)洗涤,干燥,得到化合物4-氨基-7-甲基-6-(4-硝基苯基)-7h-吡咯并[2,3-d]嘧啶(371g,产率75%)。lc-ms(esi):m/z=269.3[m+h]+
16、步骤f
17、将化合物4-氨基-7-甲基-6-(4-硝基苯基)-7h-吡咯并[2,3-d]嘧啶(18.9g,70mmol),n-碘代丁二酰亚胺溶于400mldmf中,在室温黑暗环境中反应过夜,蒸干溶剂。将残留物悬浮于10%na2so3热溶液中,过滤,用热水洗涤两次,然后从乙醇中结晶得到化合物4-氨基-5-碘-7-甲基-6-(4-硝基苯基)-7h-吡咯并[2,3-d]嘧啶(27.7g,产率100%)。lc-ms(esi):m/z=395.2[m+h]+
18、步骤g
19、在一个可密封的反应瓶中装入4-氨基-5-碘-7-甲基-6-(4-硝基苯基)-7h-吡咯并[2,3-d]嘧啶(122mg,0.31mmol)、2-(2-氟-4-(4,4,5,5-四甲基-1,3,2-二氧代硼烷-2-基)苯氧基)-5-甲基嘧啶(122mg,0.373mmol),加入pd(dtbpf)cl2(20.1mg,0.031mmol)、csf(240mg,0.930mmol)、dmf(4ml)、h2o(0.5ml)和搅拌棒,抽换n2三次,混合物在90℃下搅拌反应2h。反应混合物在真空中浓缩。所得粗物质通过tlc纯化,真空浓缩后,得到化合物5-(2-氟-4-((4-甲基嘧啶-2-基)氧基)苯基)-7-甲基-6-(4-硝基苯基)-7h-吡咯并[2,3-d]嘧啶-4-胺(8.8mg,产率6%)。lc-ms(esi):m/z=471.5[m+h]+
20、步骤h
21、在氩气条件下将5-(2-氟-4-((4-甲基嘧啶-2-基)氧基)苯基)-7-甲基-6-(4-硝基苯基)-7h-吡咯并[2,3-d]嘧啶-4-胺(0.2g,0.45mmol)和三乙基硅烷(0.45mmol)溶于乙醇(5ml)中,加入催化量的氯化钯(ii)(10mol%),将所得的混合物搅拌反应2h,蒸发溶剂,然后加水倾析,水相用二乙醚萃取,有机相经无水硫酸钠干燥,减压蒸干。残余物通过硅胶柱层析纯化得到5-(2-氟-4-((4-甲基嘧啶-2-基)氧基)苯基)-7-甲基-6-(4-胺基苯基)-7h-吡咯并[2,3-d]嘧啶-4-胺(0.14g,产率70%)。lc-ms(esi):m/z=441.5[m+h]+
22、步骤i
23、将5-(2-氟-4-((4-甲基嘧啶-2-基)氧基)苯基)-7-甲基-6-(4-胺基苯基)-7h-吡咯并[2,3-d]嘧啶-4-胺(4.4g,10mmol)和甲基丙烯酰氯(1.06ml,11mmol)溶于thf(100ml)中在0℃氩气环境下搅拌,并用碳酸钾(1.38g 10mmol)处理。反应混合物搅拌2h,然后加入2m hcl并用乙酸乙酯萃取。水层进一步用乙酸乙酯(2×50ml)萃取,合并的有机相在无水硫酸钠干燥,通过旋转蒸发去除溶剂,粗产物经柱层析纯化得到化合物rly-4008(4.1g,产率80%);lc-ms(esi):m/z=509.5[m+h]+.1h nmr(400mhz,dmso-d6)δ1.95(s,3h),2.42(s,3h),3.59(s,3h),5.54(s,1h),5.80(s,1h),5.99(s,2h),7.09(d,1h),7.18(d,2h),7.26-7.50(m,3h),7.75(d,2h),8.21(s,1h),8.47(d,1h),9.94(s,1h).
24、实施例2
25、
26、用与实施例1步骤a、b类似的方法得到化合物4-羟基-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶
27、步骤c
28、将化合物4-羟基-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(17.9g,70mmol),n-碘代丁二酰亚胺溶于400ml dmf中,在室温黑暗环境中反应过夜,蒸干溶剂。将残留物悬浮于10%na2so3热溶液中,过滤,用热水洗涤两次,然后从乙醇中结晶得到化合物4-羟基-5-碘-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(28g,产率100%)。lc-ms(esi):m/z=400.6[m+h]+
29、步骤d
30、将化合物4-羟基-5-碘-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(0.78g,2.03mmol)和三氯氧磷(10ml)的混合物回流反应3h,浓缩悬浮液,除去氧氯化磷,残留物用乙酸乙酯稀释,有机层用饱和nahco3水溶液洗涤无水硫酸钠干燥,减压蒸干,得到化合物4-氯-5-碘-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(0.16g,产率20%)。lc-ms(esi):m/z=400.6[m+h]+
31、步骤e
32、将化合物4-氯-5-碘-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(260mg,0.65mmol)和碳酸铯(319mg,0.980mmol)溶于无水dmf(2ml)中,在30min内加入碘甲烷(0.65ml,1.31mmol,2m在叔丁基甲基醚中),溶液在室温下搅拌反应90min。用h2o(50ml)淬灭反应,并用etoac(2×30ml)萃取。合并的有机相用饱和nahco3(15ml)和饱和nacl(20ml)洗涤,用无水na2so4干燥,过滤并浓缩。残余物通过硅胶柱层析纯化,得到化合物4-氯-5-碘-7-甲基-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(215mg,产率80%)。lc-ms(esi):m/z=414.6[m+h]+
33、步骤f
34、将化合物4-氯-5-碘-7-甲基-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(762g,1.84mol)悬浮于在氨水(30%h20中,3.63l)中,在压力容器中120℃搅拌下反应18h,冷却到20℃,过滤,用h20(1.80l)和甲醇(900ml)洗涤,干燥,得到化合物4-氨基-5-碘-7-甲基-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(545g,产率75%)。lc-ms(esi):m/z=395.2[m+h]+
35、接下来的三步用与实施例1中同样的方法制备得到目标化合物rly-4008。
36、实施例3
37、
38、用与实施例1步骤a、b类似的方法得到化合物4-氯-6-(4-硝基苯基)-7h-吡咯并[2,3-d]嘧啶
39、步骤c
40、将化合物4-羟基-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(0.52g,2.03毫摩尔)和三氯氧磷(10ml)的混合物回流反应3小时,浓缩悬浮液,除去氧氯化磷,残留物用乙酸乙酯稀释,有机层用饱和nahco3水溶液洗涤无水硫酸钠干燥,减压蒸干,得到化合物4-氯-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(0.11g,产率20%)。lc-ms(esi):m/z=274.7[m+h]+
41、步骤d
42、将化合物4-氯-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(19.2g,70mmol),n-碘代丁二酰亚胺溶于400ml dmf中,在室温黑暗环境中反应过夜,蒸干溶剂。将残留物悬浮于10%na2so3热溶液中,过滤,用热水洗涤两次,然后从乙醇中结晶得到化合物4-氯-5-碘-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(27.5g,产率98%)。lc-ms(esi):m/z=400.6[m+h]+
43、步骤e
44、将化合物4-氯-5-碘-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(260mg,0.65mmol)和碳酸铯(319mg,0.980mmol)溶于无水dmf(2ml)中,在30分钟内加入碘甲烷(0.65ml,1.31mmol,2m在叔丁基甲基醚中),溶液在室温下搅拌反应90分钟。用h2o(50ml)淬灭反应,并用etoac(2×30ml)萃取。合并的有机相用饱和nahco3(15ml)和饱和nacl(20ml)洗涤,用无水na2so4干燥,过滤并浓缩。残余物通过硅胶柱层析纯化,得到化合物4-氯-5-碘-7-甲基-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(238mg,产率88%)。lc-ms(esi):m/z=414.6[m+h]+
45、步骤f
46、将化合物4-氯-5-碘-7-甲基-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(762g,1.84mol)悬浮于在氨水(30%,h2o中,3.63l)中,在压力容器中搅拌下120℃反应18h,冷却到20℃,过滤,用h20(1.80l)和甲醇(900ml)洗涤,干燥,得到化合物4-氨基-5-碘-7-甲基-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(545g,产率75%)。lc-ms(esi):m/z=395.2[m+h]+
47、接下来的三步用与实施例1中同样的方法制备得到目标化合物rly-4008。
48、实施例4
49、
50、用与实施例1步骤a、b、c、d类似的方法得到化合物4-氯-7-甲基-6-(4-硝基苯基)-7h-吡咯并[2,3-d]嘧啶
51、步骤e
52、将化合物4-氯-7-甲基-6-(4-硝基苯基)-7h-吡咯并[2,3-d]嘧啶(20.21g,70mmol),n-碘代丁二酰亚胺溶于400mldmf中,在室温黑暗环境中反应过夜,蒸干溶剂。将残留物悬浮于10%na2so3热溶液中,过滤,用热水洗涤两次,然后从乙醇中结晶得到化合物4-氯-5-碘-7-甲基-6-(4-硝基苯基)-7h-吡咯并[2,3-d]嘧啶(27.65g,产率75%)。lc-ms(esi):m/z=414.6[m+h]+
53、步骤f
54、将化合物4-氯-5-碘-7-甲基-6-(4-硝基苯基)-7h-吡咯并[2,3-d]嘧啶(763g,1.84mol)悬浮于在氨水(30%,h20,3.63l)中,在压力容器中搅拌下120℃反应18h,冷却到20℃,过滤,用h20(1.80l)和甲醇(900ml)洗涤,干燥,得到化合物4-氨基-5-碘-7-甲基-6-(4-硝基-苯基)-7h-吡咯并[2,3-d]嘧啶(545g,产率75%)。lc-ms(esi):m/z=395.2[m+h]+。
55、接下来的三步用与实施例1中同样的方法制备得到目标化合物rly-4008。
56、上述实例仅用于说明本发明的实施方式,但本发明不仅仅局限于上述实例。在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内,本发明要求保护范围由权利要求书及其等效物界定。
- 还没有人留言评论。精彩留言会获得点赞!