一种脂质修饰核酸适体偶联药物及其制备方法
本发明属于生物,具体而言,涉及一种脂质修饰核酸适体偶联药物及其制备方法。
背景技术:
1、核酸适体是经指数富集配体系统进化(selex)技术筛选出的能与靶标高特异性、高选择性结合的单链dna或rna。核酸适体具有与特异性高、亲和力强、易于化学合成及修饰等特点,在分子诊断、荧光成像、靶向治疗等领域有广泛的应用前景,因此核酸适体又被称为化学家的抗体。以核酸适体作为靶分子替代单克隆抗体,通过连接子将核酸适体与小分子药物偶联,合成得到核酸适体偶联药物(aptamer drug conjugates,apdc)。apdc体系显示出了对肿瘤细胞较高的靶向性,而且几乎不与正常细胞结合,因此可极大地提高药效,并降低药物副作用。
2、然而apdc在体内应用时仍存在循环时间短、容易被肾脏代谢和核酸酶剪切的问题,迫切需要一种通用技术手段来提升在血液中的循环半衰期及稳定性。血清中含有大量的白蛋白,约为40mg/ml,且白蛋白的循环半衰期为20天,作为生物体内内源性蛋白具有良好的水溶性、生物兼容性。白蛋白已被广泛用于抗肿瘤药物的递送,例如抗癌药物紫杉醇,已被设计与白蛋白结合,降低毒性,提升水溶性,在多种动物模型上都有显著的疗效。疏水性的物质如脂质、胆固醇可以与白蛋白的疏水空腔发生非共价结合,借助白蛋白的转运。
3、公开号为cn104606127b的专利提供了一种载铂类药物的白蛋白纳米粒,并与核酸适配体交联,然而该方法药物是通过包裹的形式与白蛋白形成复合物,在体内生理环境中应用时存在药物泄露的风险。同时核酸适体与白蛋白通过共价偶联,反应过程可能造成白蛋白聚集等问题。
4、因此急需开发一种新型的核酸适体偶联药物,其能与体内的白蛋白便捷结合,从而借助白蛋白提高核酸适体偶联药物体内循环时间,解决核酸适体偶联药物面临的半衰期短、易被肾脏快速清除,同时在给药后血液循环中容易被核酸酶降解的问题,实现药物的安全靶向给药。
技术实现思路
1、为解决上述问题,本发明提供了脂质修饰核酸适体偶联药物及其制备方法,通过对核酸适体进行脂质修饰,再偶联药物,实现apdc与体内血清白蛋白的非共价结合,从而以血清中白蛋白为载体提升apdc在血液中的循环时间,实现其对肿瘤更好的靶向富集和治疗效果。与现有核酸适体偶联药物相比,脂质修饰核酸适体偶联药物具有更长的体内循环时间和稳定性,制备流程简单方便,具有极高的临床转化价值。
2、一方面,本发明提供了一种脂质修饰的核酸适体,所述核酸适体的一端含有脂质修饰,所述核酸适体通过脂质修饰可以与白蛋白结合。
3、本发明通过核酸固相合成,将能与白蛋白非共价结合的脂质高效修饰在核酸适体末端,制得脂质修饰的核酸适体,采用该脂质修饰的核酸适体,并在另一端偶联药物制得脂质修饰的apdc。经注射进入生物体内,由于脂质分子具有插入白蛋白的性质,脂质修饰的apdc遇到生物体内的白蛋白能迅速与之结合,从而以白蛋白为载体进行药物递送,利用核酸适体靶向肿瘤,并能显著提高apdc在生物体内的稳定性及循环时间,并具有较高的抗肿瘤效率。
4、进一步地,所述脂质修饰中的脂质包括芳香基脂质、或含有至少一条10~20个碳原子的饱和或不饱和烷烃脂质、胆固醇、胆固醇类似物中的任意一种或多种。
5、在一些方式中,所述脂质修饰为采用合成亚磷酰胺单体后的脂质进行修饰。
6、进一步地,所述核酸适体靶向以下任意一种或多种抗原:cd5、cd19、cd20、cd25、cd37、cd30、cd33、cd45、campath-1、hld-dr、cea、tag-72、epcam、muc1、muc15、folate-binding protein、a33、g250、psma、ferritin、gd2、gd3、gm2、leg、ca-125、ca19-9、epidermal growth factor、p185her2、il-2receptor、tenascin、a metalloproteinase、endosialin、vascular endothelial growth factor、avb3、wt1、lmp2、hpv e6、hpv e7、egfrviii、her-2/neu、mage a3、p53 nonmutant、ny-eso-1、melana/mart1、ras mutant、gp100、p53 mutant、pr1、bcr-abl、tyrosinase、survivin、psa、htert、a sarcomatranslocation breakpoint fusion protein、epha2、pap、ml-iap、afp、erg、na17、pax3、alk、androgen receptor、cyclin bl、polysialic acid、mycn、rhoc、trp-2、fucosyl gm1、msln、psca、mage al、mage-a3、sle、cyp1b1、plav1、gm3、boris、tn、globoh、etv6-aml、ny-br-1、rgs5、sart3、stn、carbonic anhydrase ix,pax5、oy-tesl sperm protein 17、lck、hmwmaa、akap-4、55x2、xage 1、b7h3、legumain、tie 3、page4、vegfr2、mad-ct-1、pdgfr-b、mad-ct-2、ror2、cmet、her3、epcam、ca6、napi2b、trop2、cldn18.2、fap、ron、ly6e、fra、dll3、ptk7、liv1、ror1、fos-related antigen 1、vegfr、endoglin、pd-l1、cd204、cd206、cd301、vtcn1、vista。
7、可以理解的是,任何序列的核酸适体都可以通过本发明提供的方法进行脂质修饰,从而构建能够与白蛋白非共价结合的脂质修饰核酸适体偶联药物。
8、在一些方式中,所述脂质通过设计合成为可用于核酸固相合成的亚磷酰胺单体,依据核酸固相合成原理,在核酸适体的5’端进行修饰,偶联药物位于核酸适体的3’端。
9、另一方面,本发明提供了一种脂质修饰核酸适体偶联药物,基于如上所述的脂质修饰的核酸适体,所述核酸适体的另一端与药物偶联,所述药物包括澳瑞他汀e、dxd、伊西替康、或与澳瑞他汀e、dxd或伊西替康具有相同或相似母核结构的衍生物中的任意一种或多种。
10、进一步地,所述脂质修饰位于核酸适体的5’端,药物位于核酸适体的3’端。
11、再一方面,本发明提供了一种脂质修饰核酸适体偶联药物的制备方法,所述方法包括以下步骤:
12、(1)对核酸适体的5’端进行脂质修饰;
13、(2)对核酸适体的3’端进行药物偶联。
14、进一步地,步骤(1)所述脂质修饰的方法包括固相合成、巯基-马来酰亚胺反应、叠氮-炔烃反应、氨基-羧基反应中的任意一种或多种;步骤(2)所述药物偶联的方法包括巯基-马来酰亚胺反应、叠氮-炔烃反应、氨基-羧基反应中的任意一种或多种。
15、可以理解的是,任何一种能使核酸适体的一端带上脂质修饰的方法,都可以用于制备本发明提供的脂质修饰核酸适体偶联药物。
16、任何一种能使核酸适体的一端偶联上药物的方法,都可以用于制备本发明提供的脂质修饰核酸适体偶联药物。
17、进一步地,步骤(1)所述脂质修饰的方法为:采用合成为亚磷酰胺单体后的脂质,经固相合成的方法,对核酸适体的5’端进行脂质修饰;步骤(2)所述药物偶联的方法为:在固相合成中对核酸适体的3’端进行巯基修饰,再经巯基-马来酰亚胺反应的方法,在核酸适体的3’端进行偶联药物。
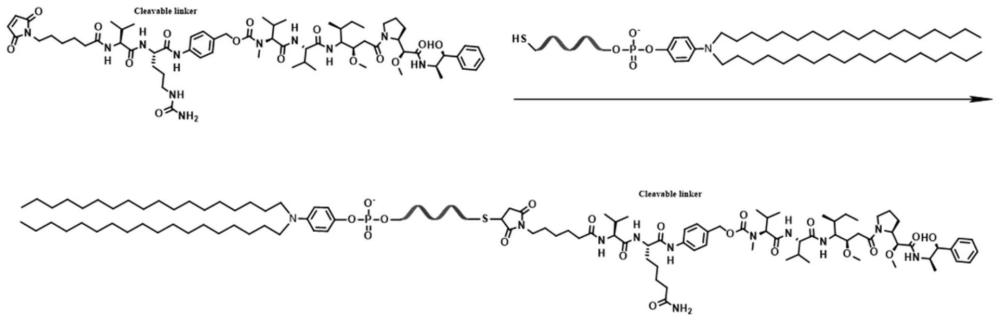
18、本发明优选采用核酸固相合成仪经固相合成的方法进行核酸适体的高效修饰,相比于巯基-马来酰亚胺反应、叠氮-炔烃反应、氨基-羧基反应等其他方法,固相合成的方法具有位点可控、高效、且可与上述反应官能团同时修饰在一条核酸适体上。
19、本发明优选采用巯基-马来酰亚胺反应的方法进行药物偶联,其原因是在具有高反应效率的前提下,含有马来酰亚胺基团的药物分子已经被广泛应用于抗体偶联药物等偶联药物的工业生产,原料成本较其他修饰低。
20、在一些方式中,步骤(2)所述药物偶联的药物为含有连接子的细胞毒药物,如一甲基澳瑞他汀e及其结构类似物、伊西替康及其结构类似物等药物。
21、在一些方式中,所述含连接子的细胞毒药物的用量为1-100当量(1当量是指摩尔量比为1:1),优选50当量;作为本发明的实施方案之一,溶剂的量为0.1-50ml,优选25ml。
22、在一些方式中,步骤(1)或步骤(2)所述反应中,溶解脂质修饰核酸适体的溶液为去离子水、三乙基乙酸铵缓冲液、磷酸盐缓冲液或它们的任意组合物。
23、在一些方式中,步骤(2)中,反应溶剂为0.1m三乙基乙酸铵缓冲液、磷酸盐缓冲液、去离子水、乙腈、二甲基亚砜、二甲基甲酰胺或它们的任意组合。
24、在一些方式中,步骤(2)中,反应温度保持在4-40℃,优选37℃,反应时间为2-12小时,优选4h。
25、进一步地,步骤(2)所述对核酸适体的另一端进行药物偶联的反应式如式(ⅰ)或式(ⅱ)所示:
26、
27、式(ⅰ)(lipid-apdc)
28、
29、式(ⅱ)(chol-apdc)
30、在一些方式中,步骤(2)中,脂质修饰核酸适体偶联药物产物还需进行分离纯化,使用高效液相色谱、疏水作用色谱、离子交换色谱中的一种进行分离纯化。
31、再一方面,本发明提供了如上所述的核酸适体或脂质修饰核酸适体偶联药物用于制备靶向治疗肿瘤的试剂的用途。
32、在一些方式中,所述肿瘤为恶性肿瘤细胞,包括ncl-h1975、ht-29等核酸适体靶标高表达细胞系。
33、本发明具有以下有益效果:
34、(1)通过脂质修饰核酸适体,在不影响与靶标结合的情况下,使其能与白蛋白的疏水口袋结合,借助白蛋白的转运提升核酸适体在血液循环中的稳定性;
35、(2)通过一步反应偶联药物——微管蛋白抑制vc-mmae剂,构建了能借助白蛋白转运的核酸适体靶标靶向的脂质核酸适体偶联药物lipid-apdc或chol-apdc;
36、(3)提高核酸适体偶联药物的稳定性及循环时间,并具有较高的抗肿瘤效率,用于核酸适体靶标高表达细胞及高表达肿瘤模型的靶向治疗,lipid-apdc细胞毒性ic50低至74.7nm,动物模型肿瘤抑制效果达75.73%以上,抑制了核酸适体靶标高表达细胞及高表达肿瘤的增殖。
- 还没有人留言评论。精彩留言会获得点赞!