一种NF1基因检测试剂及检测方法与流程
本发明属于基因检测,具体涉及一种nf1基因检测试剂及检测方法。
背景技术:
1、神经纤维蛋白(homo sapiens neurofibromin 1-nf1)的功能异常会导致一种常染色体显性疾病—1型神经纤维瘤病(nf1;omim 613113),发病率高达1:2500~3000,不受种族的影响(huson等人,1989;gutmann等人,1997;lammert等人,2005;evans等人,2010)。nf1特点是多发性咖啡斑(cafe-au-lai macule,calm)和皮肤神经纤维瘤(ferner等人,2007)。患者符合以下两个或两个以上特征,则可在临床上诊断为nf1(修订后的诊断标准,2021):六个或六个以上的牛奶咖啡斑(青春期前大直径>5 mm或成人>15 mm);两个或多个任何类型的神经纤维瘤或一个丛状神经纤维瘤;腋窝或腹股沟区域有雀斑;视神经胶质瘤;两个或多个虹膜-利希结节(虹膜错构瘤);骨损伤(蝶骨发育不良或长骨皮质变薄,伴有或不伴有假血栓形成);一级亲属中有确诊为nf1;以及nf1基因在白细胞中具有50%的变异。
2、人的nf1基因位于17q11.2中。它在基因组上跨越约300kb(gca_000001405.29)长度,包含57个组成外显子和3个可变剪切体,编码11至13kb的mrna(seq nm_000267)(cawthon等人,1990;viskochil等人,1990年;wallace等人,1990,viskochill等人,1998年)。无论是在基础科学还是在临床诊断中,nf1的基因检测都意义重大。但是,在整个人类基因组中,nf1存在有多个同源伪基因样序列(purandare等人,1995年;luijten等人,2000年;yu等人,2005年;messiaen等人,2008年),同时,nf1基因dna序列长,突变类型多,有相当部分的异常选择性剪接并且未发现变异热点,这些特点使nf1基因变异的检测面临挑战(griffiths等人,2007年;messiaen等人,2008年)。因此,nf1的基因检测需要能区分同源序列并尽可能覆盖多种变异。
3、到目前为止,临床已经应用多种检测技术进行nf1的变异检测。变性高效液相色谱(dhplc)和gdna/cdna sanger法测序用于鉴定点变异;荧光原位杂交(fish)、定量聚合酶链反应(qpcr)和多重连接依赖性探针扩增(mlpa)用于检测基因内以及整个基因拷贝数变异。两种或多种技术的组合是目前nf1变异检测的策略(griffiths等人,2006;valero等人,2011;ko等人,2013;sabbagh等人,2013年;maruoka等人,2014年;zhu等人,2016年;tspi等人,2018年;milla等人,2018)。然而,传统的检测策略,复杂度高,周期长,敏感性差,在临床上,患者需要更简单快速且全面的nf1基因检测方案。
4、专利cn 116083560 a公开了一种用于着床前胚胎nf1基因检测的试剂盒,该试剂盒包括设计的n个引物对中的一个或更多个引物对,所述n为大于2的自然数,其中第2对引物的上游引物位于第1对引物的下游引物的上游或与之重叠,依此类推,第n对引物的上游引物位于第n-1对引物的下游引物的下游或与之重叠,即所述n个引物对扩增的序列涵盖了nf1基因所有外显子的完整cds区,且n个引物对的每个引物对的上游引物和下游引物分别设计在所述目标基因的不同外显子区域,从而实现了从mrna水平进行基因序列的扩增,避免假基因和内含子的干扰,覆盖全部的cds区。但该专利采用的是sanger一代测序技术,其引物对扩增的序列只涵盖nf1基因所有外显子的完整cds区,只能检测外显子区域的点突变,无法检测因内含子变异导致的异常选择性剪接;同时需明确致病位点,再进行检测,无法对未明确致病位点的nf1患者进行临床诊断。
技术实现思路
1、针对以上现有技术存在的缺点和不足之处,本发明的首要目的在于提供一种nf1基因检测试剂。
2、本发明的另一目的在于提供一种nf1基因检测方法。
3、本发明的检测试剂及检测方法通过多对引物对患者的nf1 cdna全长进行扩增,随后进行二代测序,并对测序数据进行点变异和选择性剪接分析,不仅可对未明确致病位点的nf1患者进行临床诊断,还可检测因内含子变异导致的异常选择性剪接,可以准确、快速地检测出nf1基因多种变异,具有灵敏、可行、快速和相对经济的优点。并可进一步使用外显子捕获技术对nf1 cdna进行ngs测序二次验证,可提高检测准确性。
4、本发明目的通过以下技术方案实现:
5、一种nf1基因检测试剂,包括如下六对引物组:
6、nf1-3-f1:tgggagcctgcactccacag(seq id no:1),
7、nf1-3-r1:agcagagcctccattgcttcct(seq id no:2);
8、nf1-3-f2:ccgcattggattggtggcctaa(seq id no:3),
9、nf1-3-r2:agggcacaaaggaagccagt(seq id no:4);
10、nf1-3-f3:cattgagcatcccactgcaggaa(seq id no:5),
11、nf1-3-r3:caccaatctctcaaaccgatcagcc(seq id no:6);
12、nf1-3-f4:atgttcgtgtgcttgggaatatggt(seq id no:7),
13、nf1-3-r4:gccctggtttgcaatggttaaggt(seq id no:8);
14、nf1-3-f5:taactgtaactcctgggtcag(seq id no:9),
15、nf1-3-r5:tatacggagactatctaaagtatgcag(seq id no:10);
16、nf1-3-f6:tggcattagcaaagtcaagtc(seq id no:11),
17、nf1-3-r6:ggtcactagtgaagagcccatgt(seq id no:12)。
18、进一步地,所述nf1基因检测试剂还包括pcr缓冲液、dntp、dscdna、dna聚合酶和ddh2o。
19、进一步优选地,所述pcr缓冲液为5× prime star gxl buffer。
20、进一步优选地,所述dna聚合酶为prime star gxl dna polymerase。
21、进一步优选地,所述nf1基因检测试剂具体成分组成如下:
22、5× prime star gxl buffer 5μl;
23、2.5μm dntp 2μl;
24、10µm 混合引物组 0.875μl;
25、dscdna(20ng) 1μl;
26、 prime star gxl dna polymerase 1μl;
27、ddh2o 15.125μl。
28、一种nf1基因检测方法,包括如下步骤:
29、(1)提取待测样本rna反转录合成双链cdna;
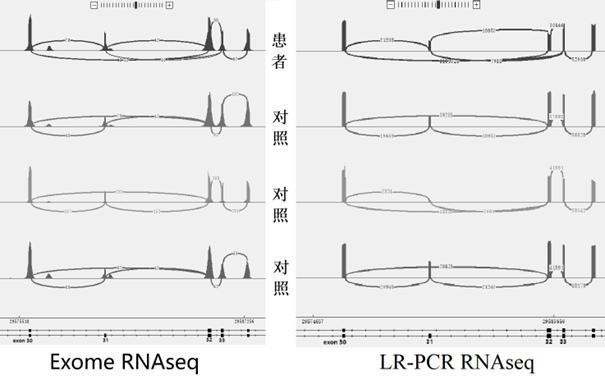
30、(2)采用上述nf1基因检测试剂对步骤(1)的双链cdna进行长片段pcr扩增后打断建库,二代测序后判断是否存在nf1基因变异。
31、进一步地,步骤(1)中所述rna的提取步骤如下:
32、将待测血液样本经离心取沉淀加入细胞裂解液孵育,所得溶液经过离心柱层析纯化与洗脱,得到rna。
33、进一步地,步骤(2)中所述pcr扩增条件如下:
34、94℃预变性1min,1循环;98℃变性10s,61℃退火10s,68℃延伸20s,40循环;72℃延伸5min,1循环。
35、进一步地,所述检测方法还包括对步骤(1)的双链cdna打断建库,对nf1外显子区域进行探针杂交捕获洗脱,二代测序验证是否存在nf1基因变异。
36、进一步地,所述nf1基因检测方法还包括生信分析与报告出具,所述生信分析与报告出具流程如下:
37、(1)使用nextgene软件,对lr-pcr扩增系统的pcr测序数据以及exome-rna测序系统的外显子组测序数据进行数据转化、参考序列比对、snv/indel变异识别和输出等,根据测序深度、read balance、maf(mutant allele frequency)、是否为exon区域变异等数值进行snv/indel的筛选,并比较pcr测序数据和外显子组测序数据在检测编码区snv/indel方面的异同;对识别出的snv/indel进行变异注释,根据人群频率、保守性分析、功能预测、公开数据(clinvar、hgmd、pubmed等)、家系分析等参数,结合患者的临床症状,进行snv/indel的致病性评级;
38、(2)根据步骤(1)识别出的snv/indel数据,通过maf值判断这些snv/indel是纯合状态还是杂合状态,如存在杂合位点,则判断nf1不存在单等位基因表达(mono allelicexpression);如果存在罕见的纯合snv/indel或多个纯合snv/indel位点,则需考虑nf1基因可能存在单等位基因表达(mono allelic expression,mae);
39、(3)使用nextgene软件,对lr-pcr扩增系统的pcr测序数据以及exome-rna测序系统的外显子组测序数据进行数据转化、参考序列比对、结构变异的识别和输出等,根据测序深度、read balance、maf等数值进行结构变异的识别,并比较pcr测序数据和外显子组测序数据在检测结构变异的异同;对识别的结构变异与正常对照样本进行比较,过滤掉正常的选择性剪接位点,并对罕见的选择性剪接进行注释;
40、(4)根据步骤(2)和(3)的分析数据,结合患者的临床信息,出具nf1 rnaseq检测分析报告。报告的主要内容包括患者基本临床信息、检测方法、snv/indel检测结果、单等位基因检测结果和检测结论等,并对阳性变异进行致病性的评级。
41、与现有技术相比,本发明的有益效果是:
42、(1)国际上对于nf1 mrna的检测,是对nf1 cdna分子进行短片段扩增(400-500bp),后续进行sanger法测序。这种方法的缺点一是工作量大,需要超过20对引物。二是由于引物数量多,会增加引物区域的变异导致的单染色体扩增而出现假阴性,降低了检测敏感性。三是由于产物片段短,而无法检测跨越产物的剪切变异。四是sanger法测序,对于低于30%的snv/indel嵌合体以及剪切变异敏感性低。基于上述问题,本发明采用长片段扩增技术(lr-pcr),使用6对引物对nf1基因进行扩增,pcr产物包含nf1全部编码区(约9k),产物间彼此包含300-500bp左右的重叠区域,对全部pcr产物混合进行ngs测序。其优点,一是工作量降低;二是引物数量少,从而减少引物区域变异造成的扩增失败或单染色体扩增的假阴性。三是,pcr产物相对较长,可以检测到较长的剪切变异,同时避免高度同源序列的干扰。四是使用ngs检测的测序深度高,对于高于5%的snv/indel嵌合,以及剪切变异,有更高的检测敏感性。
43、(2)本发明通过多对引物对患者的nf1 cdna全长进行扩增,随后进行二代测序,并对测序数据进行点变异和选择性剪接分析,不仅可对未明确致病位点的nf1患者进行临床诊断,还可检测因内含子变异导致的异常选择性剪接。
44、(3)本发明检测方法通过进一步采用exome rnaseq(外显子区域捕获测序)对双链cdna进行打断建库,随后进行临床外显子cds区域的捕获测序和数据分析,是对lr-pcrrnaseq(rna长片段pcr扩增测序)的一种补充。使用捕获测序技术,其优点,一是可以避免pcr引物区域变异导致的假阴性,二是可以检测nf1外显子全长区域发生的剪切变异。同时,exome rnaseq的结果也是对lr-pcr rnaseq的结果的二次验证。若两者结果一致,则不需要再进行sanger法验证。
45、(4)本发明所有检测技术均基于ngs测序平台,无需其它检测方法如hplc,mlpa,以及sanger法测序等,方便快捷且成本低;本发明通过对nf1基因长片段rna分子的检测,可以排除同源基因的干扰,准确度高;本发明可检测出nf1基因多种变异类型,包括点变异,剪切变异以及单等位基因表达,敏感性高。
- 还没有人留言评论。精彩留言会获得点赞!