氨基甲酸酯类化合物及其制备方法和医药用途
本发明属于医药领域,尤其涉及一种氨基甲酸酯类化合物及其制备方法和医药用途。
背景技术:
1、泛素特异性蛋白酶(usp)是去泛素化酶家族中最大的一类,其中usp7广受关注。usp7属半胱氨酸蛋白酶,主要位于细胞核内,调控多种细胞进程(如dna损伤应答、转录、表观遗传、免疫反应和病毒感染)相关蛋白的稳定性及核内外穿梭(dna repair 2019,76,30),被认为与许多疾病的发生发展密切相关。现有技术中usp7在多种肿瘤细胞中过表达,通过去泛素化作用稳定mdm2蛋白,而mdm2作为p53蛋白的主要负调控因子,不仅直接抑制p53的转录活性,还通过泛素化作用使p53水平降低(ce11 1998,95,5)。将肿瘤细胞中usp7敲除或抑制,可降低mdm2蛋白水平、升高p53水平,进而抑制肿瘤细胞增殖(mo1 ce11 2004,6,879;nature 2004,428,6982)。此外,usp7可以通过去泛素化作用稳定foxp3蛋白,而foxp3是决定免疫抑制性的调节性t细胞(treg)分化及功能的关键性转录调控蛋白。敲除或抑制usp7可降低foxp3蛋白水平,降低肿瘤微环境中treg细胞的数量和活性,从而减少肿瘤细胞经treg介导的免疫抑制,进而增加机体的抗肿瘤免疫反应(immunity 2013,39(2),259;ebiomedicine 2016,13,99)。因此,抑制usp7有望成为治疗肿瘤及其他usp7介导的疾病(如病毒感染、炎症等)的新途径。
2、gavory等报道了以compound 4(cp4)为代表的高选择性、强效的n-酰基哌啶醇类usp7抑制剂(nat chem biol 2018,14(2),118),然而cp4及其类似物的代谢稳定性很差,严重影响了其成药性。wen等报道了以l55为代表的高选择性、强效的n-苄基哌啶醇类usp7抑制剂(eur j med chem 2020,199,112279),然而l55对人肝微粒体的代谢稳定性也较差。wen等将l55的酯基替换成环状结构后,代谢稳定性显著改善,其中氟代化合物x36具有较好的caco-2细胞透膜性和大鼠口服生物利用度,但含有nh结构片段的化合物虽然usp7抑制活性更强却具有较差的caco-2透膜性和大鼠口服生物利用度(j med chem 2022,65,16622)。目前,尚没有任何usp7抑制剂进入临床研究阶段,亟待活性强且成药性好的usp7抑制剂开展临床转化。
技术实现思路
1、发明目的:为了解决上述技术问题,本发明旨在提供一种氨基甲酸酯类化合物,其不仅对usp7具有强效抑制作用,且具有良好的caco-2细胞透膜性,有利于口服吸收,可用于制备预防或治疗usp7介导疾病的药物。
2、本发明还提供所述化合物或其药学上可接受的盐、酯、立体异构体、氘代物或溶剂化物的制备方法和医疗用途。
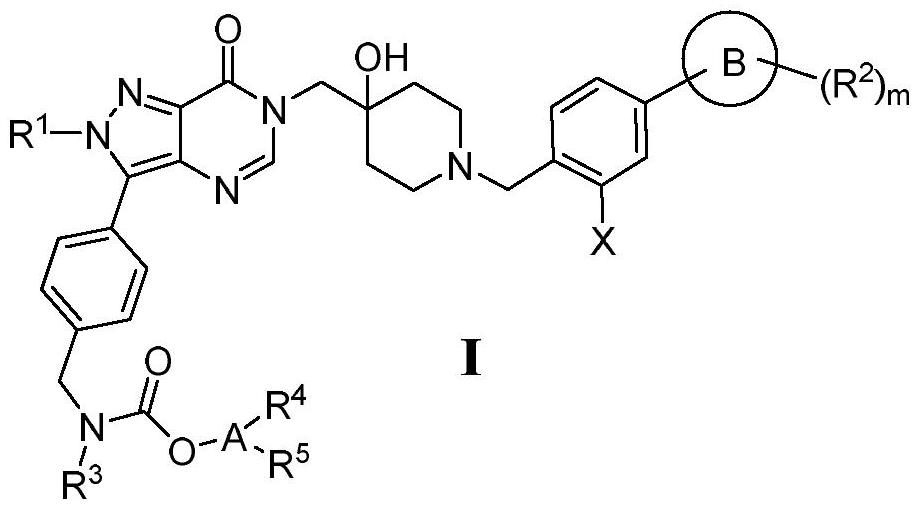
3、技术方案:为了实现上述目的,本发明如式i所示的氨基甲酸酯类化合物或其药学上可接受的盐、酯、立体异构、氘代物或溶剂化物:
4、
5、其中,r1选自h、c1-3的烷基、或者环丙基;
6、x选自f、cl、br或i;
7、环b选自五至六元的芳基或杂芳基、或者三至六元的环烷基或杂环烷基;
8、环b被m个独立选择的r2取代,m选自0、1或2,r2选自h、卤素、cn、ora、nhra、n(ra)2、nhcora、nhso2ra、coora、conhra、con(ra)2、so3ra、so2nhra、so2n(ra)2、c1-6的烷基、或者三至六元的环烷基或杂环烷基;
9、r3选自h、c1-6的烷基、或者三至四元的环烷基或杂环烷基;
10、a选自ch、五元或六元芳基或杂芳基、或者三至六元的环烷基或杂环烷基;
11、a为ch时,r4、r5各自独立地选自h、取代或非取代的c1-6的烷基、取代或非取代的三至六元的环烷基或杂环烷基、取代或非取代的五元或六元芳基或杂芳基、cn、coorb、conhrb、con(rb)2、so3rb、so2nhrb、so2n(rb)2、或者取代或非取代的烯基或炔基;
12、a为芳基、杂芳基时,r4、r5各自独立地选自h、卤素、cn、orc、nhrc、n(rc)2、nhcorc、nhso2rc、coorc、conhrc、so3rc、或者so2nhrc、so2n(rc)2;
13、ra、rb、rc各自独立地选自h、c1-6的烷基、或者三至六元的环烷基或杂环烷基;
14、以上所述杂芳基、杂环烷基指1~3个杂原子各自独立地选自n、o或s。
15、进一步地,所述环b选自如下杂环中的任意一种:
16、
17、进一步地,所述优选自如下结构片段中的任意一种:
18、
19、进一步地,所述化合物选自下列化合物任意一种,所述化合物的结构详见表1:
20、表1
21、
22、
23、
24、
25、
26、
27、
28、
29、
30、
31、
32、
33、进一步地,本发明的化合物可作为药用盐使用。
34、作为优选,所述的盐可为下列酸中至少一种的酸盐:半乳糖二酸、d-葡糖醛酸、甘油磷酸、马尿酸、羟乙磺酸、乳糖酸、马来酸、1,5-萘二磺酸、萘-2-磺酸、新戊酸、对苯二甲酸、硫氰酸、胆酸、正十二烷基硫酸、苯磺酸、柠檬酸、d-葡萄糖,乙醇酸、乳酸、苹果酸、丙二酸、扁桃酸、磷酸、丙酸、盐酸、硫酸、酒石酸、琥珀酸、甲酸、氢碘酸、氢溴酸、甲烷磺酸、烟酸、硝酸、乳清酸、草酸、苦味酸、l-焦谷氨酸、糖精酸、水杨酸、龙胆酸、对甲苯磺酸、戊酸、棕榈酸、葵二酸、硬脂酸、月桂酸、乙酸、己二酸、碳酸、苯磺酸、乙烷二磺酸、乙基琥珀酸、富马酸、3-羟基萘-2-甲酸、1-羟基萘-2-甲酸、油酸、十一碳烯酸、抗坏血酸、樟脑酸、樟脑磺酸、二氯乙酸、乙烷磺酸。
35、所述的盐也可以为与药学上可接受的金属(包括钠、钾、钙等)离子或胺(包括乙二胺、氨丁三醇等)、铵离子或胆碱形成的盐。
36、进一步地,所述合成路线如下任意一种所示:
37、
38、合成过程为:哌啶化合物与卤代化合物之间的亲核取代,或与醛在还原剂介导的条件下发生还原胺化反应,或氨基化合物与醇在缩合剂的条件下发生缩合反应。
39、其中,所述化合物或其药学上可接受的盐、酯、立体异构体、氘代物或溶剂化物在制备usp7抑制剂中的用途。
40、本发明所述化合物或其药学上可接受的盐、酯、立体异构体、氘代物或溶剂化物在制备预防或治疗usp7介导的疾病的药物中的用途。
41、进一步地,所述usp7介导的疾病为肿瘤、病毒感染性疾病、炎症性疾病或自身免疫性疾病中任意一种。
42、所述的肿瘤疾病包括但不限于:骨癌、急性骨髓性白血病、慢性骨髓性白血病、急性淋巴细胞系白血病、慢性淋巴细胞系白血病、骨髓增生性疾病、多发性骨髓瘤、骨髓增生异常综合征、霍奇金氏淋巴瘤、非霍奇金氏淋巴瘤、血管瘤、肉芽瘤、黄瘤、脑膜肉瘤、神经胶质瘤、神经母细胞瘤、星形细胞瘤、成神经管细胞瘤、室管膜瘤、生殖细胞瘤(松果体瘤)、多形性成胶质细胞瘤、少突神经胶质细胞瘤、神经鞘瘤、成视网膜细胞瘤、纤维神经瘤、肉瘤、食道癌、胃癌、胰腺癌、大肠癌、结肠癌、直肠癌、肾癌、前列腺癌、淋巴癌、睾丸癌、间质细胞癌、肺癌、肝癌、皮肤癌、噁性黑素瘤和基底细胞癌等。
43、所述的病毒感染性疾病包括但不限于以下病毒引起的感染性疾病:结核分枝杆菌感染、衣原体感染、疱疹病毒(单纯疱疹病毒)感染、腺病毒感染、乙肝病毒感染、正粘病毒感染、冠状病毒感染、细菌感染和支原体感染等。
44、所述炎症、自身免疫性疾病包括但不限于溃疡性结肠炎、克罗恩病、系统性红斑狼疮、类风湿关节炎、银屑病、多发性硬化症或白塞氏病。
45、其中,本发明提供一种药物组合物,包含通式i所示化合物或其药学上可接受的盐、酯、立体异构体、氘代物或溶剂化物和药学上可接受的载体或赋形剂。
46、进一步地,所述药物组合物是胶囊剂、散剂、片剂、颗粒剂、丸剂、注射剂、糖浆剂、口服液、吸入剂、软膏剂、栓剂或贴剂。
47、作为优选,由于本发明的化合物口服生物利用度高,所述药物组合物为口服药剂。
48、本发明化合物的各种氘代形式,即与碳原子相连的每个氢原子可以独立的被氘原子取代。
49、本发明包括本发明化合物的各种前药形式。
50、本发明的化合物可单独使用或可与其他治疗剂组合使用。
51、有益效果:与现有技术相比,本发明具有如下显著优点:
52、1、文献(j med chem 2022,65,16622)报道的含有nh结构片段的n-苄基哌啶醇类化合物具有较差的caco-2细胞透膜性和大鼠口服生物利用度。而本发明特定设计的将n-苄基哌啶醇类化合物中的nh衍生成氨基甲酸酯后显著改善了caco-2细胞透膜性和大鼠口服生物利用度,因而具有更好的成药性;
53、2、本发明提供的氨基甲酸酯类化合物或其药学上可接受的盐、酯、立体异构体或溶剂化物对usp7酶具有很强的抑制活性;
54、3、本发明提供的氨基甲酸酯类化合物或其药学上可接受的盐、酯、立体异构体、氘代物或溶剂化物,结构简单,设计巧妙,原料便宜易得,合成工艺安全、环保,易于规模化生产。
- 还没有人留言评论。精彩留言会获得点赞!