一种制备咪唑乙醇的方法与流程
本发明涉及有机合成,具体涉及一种制备咪唑乙醇的方法。
背景技术:
1、咪唑乙醇,化学名称为α-(2,4-二氯苯基)-咪唑-1-乙醇,是益康唑、奥康唑、噻康唑、硝酸咪康唑等抗真菌药物和水果保鲜剂的重要中间体,是一种用途十分广泛的化学品。
2、目前,关于咪唑乙醇的主要合成路线为以2,4,4-三氯代苯乙酮为原料,采用异丙醇铝、硼氢化钠或硼氢化钾作为还原剂将羰基还原成相应的醇,接着在强碱(氢氧化钠、甲醇钠或者钠氢等)条件下与咪唑缩合得到产物咪唑乙醇。此种合成路线非常容易产生废盐和大量的副产物,导致最终咪唑乙醇的收率较低。并且更重要的是,此条合成路线在反应后的所产生的废盐(铝盐和硼盐)等固废处理困难,对于环境存在严重污染。并且,由于以上合成路线在缩合反应中会采用强碱等试剂,同样会导致产生较多的强碱废水,进一步增加后处理的难度,同时进一步增加咪唑乙醇的制备成本,不利于咪唑乙醇的大规模生产和应用。
3、中国专利cn102180835b公开了一种制备咪唑芳香醇类衍生物的方法,该方法中虽然避免了使用强碱进行缩合反应,但是同时需要使用硼氢化钾,同样存在反应后的硼盐等固废难以处理等问题,对环境并不友好。同时,由于硼盐的存在增加了后续产品的提纯难度,需要进行多次提纯,进一步导致咪唑乙醇产品的生产成本增加。更重要的是,此合成路线同样存在多步化学反应,合成路线繁琐,进而导致咪唑乙醇的收率无法得到进一步提升,依旧无法解决目前咪唑乙醇收率低,原料转化率低且副产物量多以及后处理困难等问题。
技术实现思路
1、本发明旨在改善现有技术存在的咪唑乙醇的合成路线中,咪唑乙醇收率低,原料转化率低且副产物量多以及后处理困难,容易造成环境污染等问题。
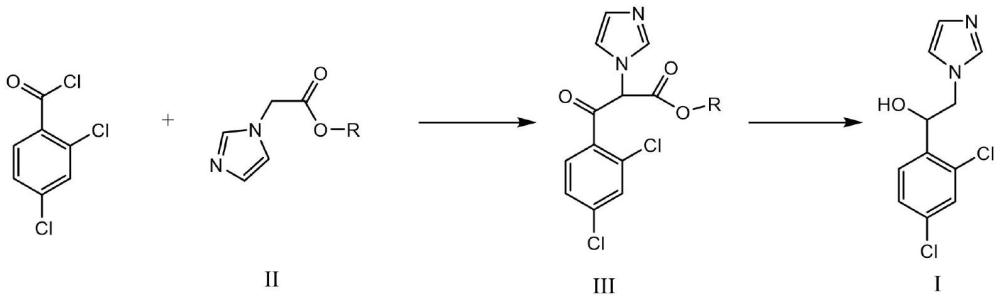
2、为了实现上述目的,本发明提供一种制备式i所示的咪唑乙醇的方法,其中所述方法包括以下步骤:
3、
4、(1)在碱的存在下,将2,4-二氯苯甲酰氯与化合物ii反应,合成式iii化合物;
5、(2)在催化剂和酸的存在下,将式iii化合物在与氢气反应,合成式i所示的咪唑乙醇;其中,所述的催化剂为钯炭或镍;所述的酸为对甲苯磺酸;
6、r选自c1-6烷基。
7、根据本发明的实施方案,r选自甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基。
8、根据本发明的实施方案,步骤(1)中,2,4-二氯苯甲酰氯与化合物ii的摩尔比为1:(1-2),优选为1:(1.1-1.5)。
9、根据本发明的实施方案,步骤(1)中,所述的碱选自氢化钠、叔丁醇钾、叔丁醇钠、三乙胺等中的一种、两种或更多种,优选为氢化钠。
10、根据本发明的实施方案,步骤(1)在碱性条件下进行反应。
11、根据本发明的实施方案,步骤(1)在有机溶剂的存在下进行反应。优选地,所述的有机溶剂选自乙二醇二甲醚、二乙二醇二甲醚、四氢呋喃、甲苯中的一种、两种或更多种。
12、根据本发明的实施方案,步骤(1)中,所述碱的用量与2,4-二氯苯甲酰氯的摩尔比为4-1:1,优选为(1.5-1):1,例如1.5:1、1.4:1、1.3:1、1.2:1、1.1:1或1:1。
13、根据本发明的实施方案,步骤(1)中,2,4-二氯苯甲酰氯以滴加的方式加入化合物ii、有机溶剂和碱的混合物中。
14、根据本发明的实施方案,步骤(1)中,先将化合物ii、有机溶剂和碱反应,再将2,4-二氯苯甲酰氯以滴加的方式加入化合物ii、有机溶剂和碱的混合物中。
15、根据本发明的实施方案,步骤(1)中,所述化合物ii、有机溶剂和碱的反应在加热条件下进行,优选回流条件下进行。
16、根据本发明的实施方案,步骤(1)中,所述化合物ii、有机溶剂和碱的反应时间为0.5-2小时,例如0.5小时、1小时、1.5小时或2小时。
17、根据本发明的实施方案,步骤(1)中,2,4-二氯苯甲酰氯的滴加时间可以为0.5-2小时,例如0.5小时、1小时、1.5小时或2小时。
18、根据本发明的实施方案,步骤(1)中,在2,4-二氯苯甲酰氯以滴加的方式加入化合物ii、有机溶剂和碱的混合物后,继续反应1-4小时,例如1、2、3或4小时。
19、根据本发明的实施方案,步骤(1)中,反应温度为50-150℃,优选为70-90℃,如80-85℃。
20、根据本发明的实施方案,步骤(1)中,将液相hplc检测反应物料中2,4-二氯苯苯甲酰氯的面积含量小于0.5%作为确定反应完毕的终点。
21、根据本发明的实施方案,步骤(1)中,反应完毕后,淬灭反应体系。例如,通过加入甲醇淬灭反应体系。
22、根据本发明的实施方案,步骤(1)中,淬灭反应体系的试剂(如甲醇)的用量可以根据残余的2,4-二氯苯苯甲酰氯的量来确定。例如,淬灭反应体系的试剂(如甲醇)的用量为2,4-二氯苯苯甲酰氯摩尔量的1%~15%,例如5%、6%、7%、8%、9%、10%、11%、12%、13%、14%或15%。
23、根据本发明的实施方案,步骤(1)中,淬灭反应体系后,减压蒸馏除去溶剂。
24、根据本发明的实施方案,步骤(1)中,处于溶剂后的残余物不经额外纯化,直接进行步骤(2)。
25、根据本发明的实施方案,步骤(2)中,基于式iii化合物的重量计,所述催化剂的用量为1-10重量%,例如1重量%、2重量%、3重量%、4重量、5重量%、6重量、7重量%、8重量%、9重量%或10重量%。
26、根据本发明的实施方案,步骤(2)中,当所述催化剂为钯碳时,其中钯碳中负载的钯的重量百分比含量为0.5~10重量%,例如0.5重量%、1重量%、2重量%、3重量%、4重量、5重量%、6重量、7重量%、8重量%、9重量%或10重量%。
27、根据本发明的实施方案,步骤(2)中,基于式iii化合物的摩尔量计,对甲苯磺酸的用量为式iii化合物摩尔量的1-15%,例如1%、2%、3%、4、5%、6%、7%、8%、9%、10%、11%、12%、13%、14%或15%。
28、根据本发明的实施方案,步骤(2)在有机溶剂的存在下进行。优选地,所述的有机溶剂选自氯苯、苯甲醚、二甲苯、dmf(n,n-二甲基甲酰胺)、dmac(n,n-二甲基乙酰胺)中的一种、两种或更多种。
29、根据本发明的实施方案,步骤(2)中,所述反应的压力为0.45-0.6mpa,如0.5mpa。
30、根据本发明的实施方案,步骤(2)中,所述反应的温度为100-200℃,例如100℃、110℃、120℃、130℃、140℃、150℃、160℃、170℃、180℃、190℃、或200℃。
31、根据本发明的实施方案,步骤(2)中,所述反应的时间为1-4小时,例如1、2、3或4小时。
32、根据本发明的实施方案,步骤(2)中,在式iii化合物与氢气反应之前,采用保护气将步骤(2)反应器中的气体置换,例如置换2-4次,如3次。
33、根据本发明的实施方案,步骤(2)中,所述的保护气为对反应呈惰性的气体,例如氮气。
34、根据本发明的实施方案,步骤(2)中,在式iii化合物与氢气反应之前,除采用保护气将步骤(2)反应器中的气体置换外,还进一步使用氢气将步骤(2)反应器中的保护气体置换,例如置换2-4次,如3次。
35、根据本发明的实施方案,步骤(2)中,将液相hplc检测反应物料中化合物iii的面积含量小于0.5%作为确定反应完毕的终点。
36、根据本发明的实施方案,步骤(2)反应完成后,过滤回收催化剂,反应液减压蒸馏回收溶剂。
37、根据本发明的实施方案,步骤(2)中,回收催化剂和溶剂后,残余物加入溶剂重结晶,干燥得到式i所示的咪唑乙醇。
38、根据本发明的实施方案,步骤(2)中,重结晶使用的溶剂为乙醇或乙醇的水溶液,例如50重量%~99重量%乙醇水溶液,如75重量%~99重量%乙醇水溶液,其实例为95重量%乙醇水溶液。
39、有益效果
40、本发明方法的反应收率高、产物纯度高、条件温和。并且,本发明方法的原料易得、成本低、安全性好,并且产生的三废少、对环境优友好,适于绿色规模化生产。
- 还没有人留言评论。精彩留言会获得点赞!