一种鉴别荞麦属植物的方法
本发明涉及一种植物鉴别方法,具体涉及一种鉴别荞麦属植物的方法。
背景技术:
1、荞麦属(fagopyrum mill),属原始花被亚纲蓼科,是一年生、多年生草本,稀半灌木,该属共有15种,广布于亚洲及欧洲。dna分子标记作为现有的先进准确性高的一项技术,可以弥补和克服传统鉴定方法上的一些缺陷和难题。但是,每一种分子标记技术都有其自身的特质和适用范围以及通用性普遍较差的问题。目前,dna条形码技术可以利用相对较短的标准dna片段对物种进行快速准确鉴定,在学术界被广泛使用,在物种鉴定领域也得到了有效地应用,可快速、准确地鉴别植物种类(li xw etal.plant dna barcoding:from geneto genome[j].biol rev,2015,90:157-166)。该技术在中药材鉴定中得到了广泛的应用从而使其快速发展,在药用植物及植物源药材鉴定等方面均获得了突出成果(yang jbetal.applying plant dna barcodes toidentify species of parnassia(parnassiaceae)[j].mol ecol resour,2012,12:267-275),加快了中药鉴定标准化的进程。
2、分子标记的概念以个体间遗传物质内核苷酸序列变异为基础的遗传标记。与其他几种遗传标记——形态学标记、生物化学标记、细胞学标记相比,dna分子标记具有的优越性有:大部分分子标记为共显性,对隐性的性状的选择十分便利;基因组变异极其丰富,分子标记的数量几乎无限;不同组织的dna都可用于标记分析;分子标记揭示来自dna的变异;表现为中性,不影响目标性状的表达,与不良性状无连锁;检测手段简单快捷。dna分子标记技术已有数十种,应用于遗传育种、基因组作图、基因定位、物种亲缘关系鉴别、基因库构建、基因克隆等方面。标记技术为所用引物的核苷酸序列是随机的,其扩增的dna区域事先未知。随机引物pcr扩增的dna区段产生多态性的分子基础是模板dna扩增区段上引物结合位点的碱基序列的突变,不同来源的基因组在该区段上表现为扩增产物有无差异或扩增片段大小的差异。随机引物pcr标记表现为显性或共显性。
3、前人对荞麦多年详细的研究为荞麦的精确鉴定及其种质资源的合理开发和利用提供了重要证据。ncbi数据库有大量关于荞麦基因组的测序和分析,揭示荞麦的基因组结构、基因家族以及遗传多样性的研究也有多家报道,但目前对荞麦进行研究的样本数量相对较少,达不到较好的重复性,使得数据结论不全面不完善,这限制了我们对荞麦遗传多样性和变异性的全面了解。一些常见的荞麦较好区分,比如甜荞麦、苦荞麦、米荞、翅荞等,但不少品种较为类似,难以鉴别。因此如何克服现有技术的不足是目前分子标记领域亟需解决的问题。
技术实现思路
1、本发明的目的是提供一种鉴别荞麦属植物的方法,解决了现有技术存在难以用传统的表型特征或理化分析等方法来鉴别荞麦植物的技术问题,使用matk基因序列进行物种鉴别,以使实验数据具有足够的可靠性,从而为确保其种质资源的正确性及保障药用的安全提供重要的分子依据。
2、为了达到上述目的,本发明提供了一种鉴别荞麦属植物的方法,该方法包含:
3、(1)对待鉴定样本进行总dna提取;
4、(2)以步骤(1)提取的总dna为模板,用seq id no.1和seq id no.2所示的引物进行pcr扩增,获得扩增产物;
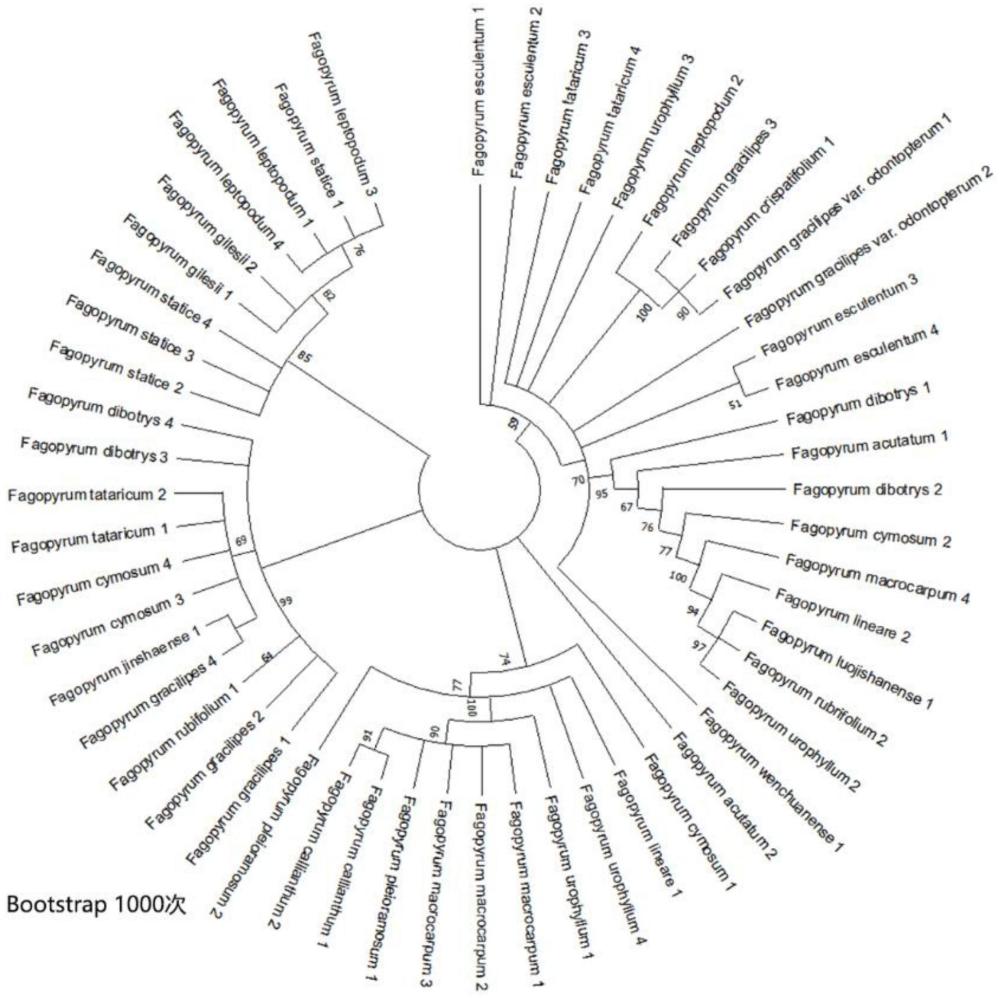
5、(3)使用codoncode aligner对扩增产物测序峰图进行校对拼接,去除低质量序列及引物区,用clustalxv2.0进行多重序列比对后,去除两端的多余序列,处理后的序列用于序列分析;使用mega11.0软件计算序列的k-2-p距离,分析比较序列的种间遗传距离,并构建系统进化树,从而通过对不同种荞麦的matk序列的聚类来区分荞麦物种。
6、优选地,所述pcr扩增的体系为:ddh2o、2×taq pcr mastermix、seq id no.1所示的引物、seq id no.2所示的引物和模板dna。
7、优选地,所述pcr扩增的体系为:ddh2o 9.0μl、2×taq pcr mastermix 12.0μl、seq id no.1所示的引物2.5μmol/l 1μl、seq id no.2所示的引物2.5μmol/l 1μl和模板dna 50ng/μl 2.0μl。
8、优选地,所述pcr扩增的程序为:95℃预变性5min;然后进入循环,每个循环95℃变性50sec,54℃退火30sec,72℃延伸60sec,共30个热循环;最后延伸73℃延伸7min;4℃冷却后取出。
9、优选地,待鉴定样本叶片按照ctab法提取总dna,使用rna酶a纯化总dna样本后,用紫外分光光度计测定260nm和280nm下的光密度值,当测得dna纯度达到od260/od280的比值在1.6~1.8之间,则进行步骤(2)pcr反应;反之,则需要重新提取dna。
10、优选地,所述荞麦属植物选自fagopyrum leptopodum、fagopyrum statice、fagopyrum rubifolium、fagopyrumpleioramosum、fagopyrum macrocarpum、fagopyrumcallianthum、fagopyrum urophyllum、fagopyrum lineare、fagopyrum cymosum、fagopyrum gilesii、fagopyrum tataricum、fagopyrum esculentum、fagopyrumdibotrys、fagopyrum gracilipes、fagopyrum gracilipes var.odontopterum、fagopyrumacutatum、fagopyrum luojishanense、fagopyrum jinshaense、fagopyrum wenchuanense、fagopyrum crispatifolium、fagopyrum dibotrys(d.don)hara.cv.lql、fagopyrumdibotrys(d.don)hara.cv.qn。
11、优选地,使用codoncode aligner对扩增产物测序峰图进行校对拼接。
12、优选地,用clustalxv2.0进行多重序列比对后,去除两端的多余序列。
13、优选地,使用mega11.0软件计算序列的k-2-p距离。
14、本发明的鉴别荞麦属植物的方法,解决了现有技术存在难以用传统的表型特征或理化分析等方法来鉴别荞麦植物的技术问题,具有以下优点:
15、本发明对四川农业大学自主栽培的2个荞麦属物种进行了测序,同时收集了20个荞麦属物种,涵盖了荞麦属大部分物种,使用matk基因序列进行物种鉴别,以使实验数据具有足够的可靠性,从而为确保其种质资源的正确性及保障药用的安全提供重要的分子依据。
16、本发明通过建立的22种荞麦属植物的dna条形码,能轻易地将各物种区分开来。通过对22种荞麦属植物的matk基因片段的pcr测序分析,在构建的系统发育树上不同物种的样本均各自聚为一支,因此可以通过对不同种荞麦的matk基因序列的聚类能准确地区分荞麦属植物,用于荞麦属植物的种间鉴别。
17、本发明利用高效自动化技术测序,即缩短了实验时间又降低了繁杂的人工测序劳作,易于对扩增样本进行标准化分析,提高了数据的可靠性。
- 还没有人留言评论。精彩留言会获得点赞!