一种体外大片段基因组DNA的合成组装方法
本发明属于合成生物学,具体涉及一种体外大片段基因组dna的合成组装方法。
背景技术:
1、合成dna的能力使研究人员能够控制dna序列的组成,从而消除了对从生物体中分离出来的自然序列的依赖,并为设计生命系统提供了一种多功能工具。随着设计复杂的代谢途径、遗传回路甚至整个生物体基因组的需求不断增加,对合成dna,特别是大片段dna的需求稳步增长。利用限制性内切酶将小片段dna连接到大分子上的方法有多种,如biobrick、golden gate和yeastfab。然而,限制性内切酶通常只产生0-4个核苷酸的悬臂,这些悬臂太短,无法为多个片段的组装提供足够的特异性和亲和力,这使得大多数组装技术只适用于生成低于10kbp的dna分子。随着所需dna序列的大小和复杂性的增加,组装的成功率急剧下降,使其难以扩展到更大的组装。
2、一些不依赖于限制性内切酶的方法,如序列和连接不依赖克隆(slic)、gibson组装或聚合酶循环组装(pca)也得到了发展。gibson组装由于其简单、高效和在最终结构中没有限制性位点或疤痕序列而被广泛采用。然而,gibson组装仅适用于组装一定长度的dna框架,通常在10-15kb左右,不适合组装整个染色体。此外,通过利用酿酒酵母的同源重组能力,已经成功组装了大小从几十到几百kb不等的合成dna结构,甚至是整个细菌基因组。近年来,科学家们利用一种名为“交换营养缺陷体渐进式整合(swap-in)”的方法,在体内构建了多条合成酵母染色体,这种方法是用合成序列迭代地替换天然序列。虽然合成染色体可以成功地组装并整合到酵母细胞中,但将组装好的染色体从酵母转移到其他生物(如哺乳动物细胞)仍然非常困难。
技术实现思路
1、为了解决现有技术中的不足,本发明的目的在于提供一种体外大片段基因组dna的合成组装方法。
2、本发明的具体技术方案如下:
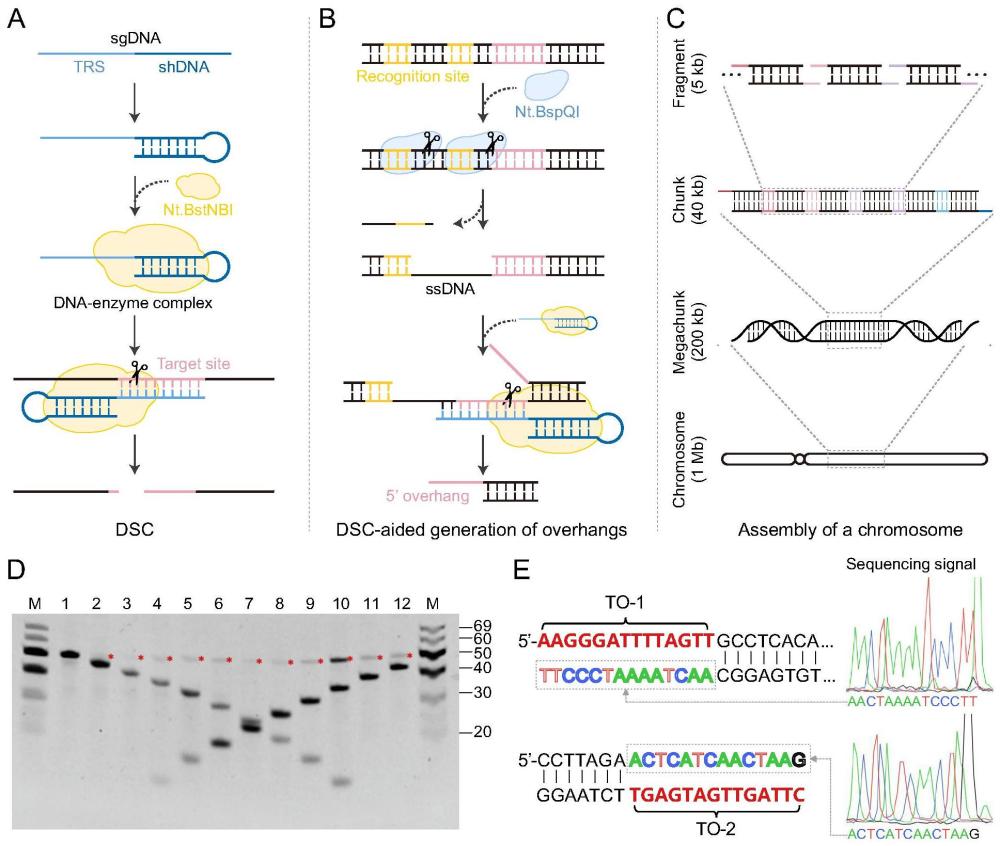
3、本发明提供一种双链dna粘性末端的生成系统在体外大片段基因组dna的合成组装中的用途,所述系统包括dna组装序列、单链引导dna、切刻内切酶a、切刻内切酶b、切刻内切酶c;
4、所述dna组装序列自5’端至3’端包括第一切刻内切酶识别位点单元、目标dna序列和第二切刻内切酶识别位点单元,所述第一切刻内切酶识别位点单元包括2或3个切刻内切酶a识别位点和位于所述切刻内切酶a识别位点之间的间隔碱基,所述第二切刻内切酶识别位点单元包括2或3个切刻内切酶b识别位点和位于所述切刻内切酶b识别位点之间的间隔碱基,所述切刻内切酶a和切刻内切酶b属于同类切刻内切酶,所述切刻内切酶a和切刻内切酶b的切割位点位于dna的上下两条链上,所述dna组装序列经切刻内切酶a和切刻内切酶b酶切处理后在dna组装序列两端分别形成单链区;
5、所述单链引导dna包括第一单链引导dna和第二单链引导dna,所述单链引导dna由靶序列的识别序列和折叠成茎环结构的短dna序列组成,所述靶序列为单链dna序列且包括所述单链区的部分序列以及其相邻目标dna序列的部分序列,该单链区的部分序列被称为第一靶序列,相邻目标dna序列的部分序列被称为第二靶序列,所述短dna序列折叠成的茎环结构具有切刻内切酶c识别位点,但缺少可被所述切刻内切酶c切割的序列,所述靶序列的识别序列与第一靶序列杂交后,第二靶序列所对应的另一条dna链被分离,靶序列的识别序列进一步与第二靶序列杂交,靶序列与识别序列杂交形成双链结构,该双链结构可被所述切刻内切酶c识别,并使第二靶序列的预定位置处于可以介由所述切刻内切酶c对单链引导dna上的识别序列的识别而被所述切刻内切酶c切割的位置,所述第一单链引导dna和第二单链引导dna的靶序列的识别序列分别与经切刻内切酶a和切刻内切酶b酶切处理后在dna组装序列两端形成的单链区的部分序列以及其相邻目标dna序列的部分序列杂交形成双链结构。
6、进一步地,所述切刻内切酶a和切刻内切酶b分别选自nt.bbvci、nt.alwi、nt.bsmai、nt.bspqi、nt.bstnbi、nt.cvipii中的一种,或者nb.bbvci、nb.bsmi、nb.bsrdi、nb.btsi中的一种;
7、优选地,所述切刻内切酶a和切刻内切酶b为同种切刻内切酶;
8、优选地,所述切刻内切酶a和切刻内切酶b为nt.bspqi;
9、优选地,所述第一切刻内切酶识别位点单元包括3个切刻内切酶a识别位点,所述第二切刻内切酶识别位点单元包括3个切刻内切酶b识别位点。
10、进一步地,所述切刻内切酶a识别位点之间的间隔碱基满足如下条件:所述切刻内切酶a的切割位点不位于其他切刻内切酶a识别序列中;
11、所述切刻内切酶b识别位点之间的间隔碱基满足如下条件:所述切刻内切酶b的切割位点不位于其他切刻内切酶b识别序列中;
12、优选地,所述切刻内切酶a识别位点之间的间隔碱基和所述切刻内切酶b识别位点之间的间隔碱基为4~10个任意排列的a、c、g或t,且间隔碱基不形成切刻内切酶识别位点。
13、进一步地,所述切刻内切酶c选自nt.bbvci、nt.alwi、nt.bsmai、nt.bspqi、nt.bstnbi、nt.cvipii、nb.bbvci、nb.bsmi、nb.bsrdi、nb.btsi中的一种;
14、优选地,所述切刻内切酶c选自nt.bstnbi。
15、进一步地,所述dna组装序列的第一切刻内切酶识别位点单元的5’端和第二切刻内切酶识别位点单元的3’端分别包括大小为500-1500bp的辅助序列;或者,将所述dna组装序列克隆至质粒中。
16、进一步地,所述单链区的长度为5-100个碱基。
17、进一步地,所述第二靶序列的长度在5-100碱基之间,优选在5-30碱基之间,更优选在10-30碱基之间。
18、进一步地,所述粘性末端的长度为5-30碱基之间,优选为5-20碱基之间。
19、进一步地,所述大片段基因组dna为酵母染色体或t7噬菌体基因组。
20、本发明还提供一种体外大片段基因组dna的合成组装方法,采用所述的系统,包括如下步骤:
21、(1)将基因组序列分成n个目标dna序列,n为正整数,制备n个dna组装序列,设计并合成每个dna组装序列的单链引导dna;
22、(2)向每个dna组装序列、与其对应的单链引导dna中加入切刻内切酶a、切刻内切酶b和切刻内切酶c,进行酶切处理,相邻目标dna序列的dna组装序列经酶切处理后产生的粘性末端互补;
23、(3)将步骤(2)酶切处理后的dna组装序列进行dna体外组装。
24、进一步地,步骤(1)中所述目标dna序列的大小为300bp-50kb;
25、优选地,步骤(1)中所述dna组装序列的制备方法为:将目标dna序列克隆至包含第一切刻内切酶识别位点单元和第二切刻内切酶识别位点单元的质粒中,且目标dna序列位于第一切刻内切酶识别位点单元和第二切刻内切酶识别位点单元之间;或者,在每个目标dna序列的两端分别插入第一切刻内切酶识别位点单元和第二切刻内切酶识别位点单元,扩增获得所述dna组装序列;
26、优选地,所述质粒为pcce,其核苷酸序列如seq id no.13所示;
27、优选地,所述dna组装序列的第一切刻内切酶识别位点单元的5’端和第二切刻内切酶识别位点单元的3’端分别包括大小为500-1500bp的辅助序列,扩增获得所述dna组装序列。
28、进一步地,步骤(2)中所述酶切处理在30-75℃下进行,优选在45-65℃下进行,更优选在50-60℃下进行;
29、优选地,步骤(2)中所述酶切处理的体系为:4μg dna组装序列,1xcutsmartbuffer,1mm dtt,体积百分比浓度8%的peg4000,3μm单链引导dna,12u切刻内切酶a、12u切刻内切酶b和12u切刻内切酶c;酶切处理的条件为在50℃孵育2h。
30、进一步地,步骤(3)中所述dna体外组装根据基因组dna片段的大小,进行多轮组装,从而得到大片段基因组dna;
31、优选地,步骤(3)中所述dna体外组装包括依次进行的杂交反应和组装反应两个步骤:
32、1)杂交反应:在杂交缓冲液中加入相邻的酶切后dna组装序列,进行杂交反应;杂交反应条件为:55±5℃,5±3min;45±5℃,10±5min;40±5℃,10±5min;35±5℃,10±5min;20±5℃,10±5min;杂交缓冲液组分为100mmnacl,20mm tris-ac,1mm edta,体积百分比浓度8%-16%的peg4000,ph7.5;
33、2)组装反应:杂交反应结束后,向杂交反应体系中加入组装缓冲液和dna连接酶;组装反应条件为:30±5℃,10±5min;50-100个循环×(30±5℃,1±0.5min;42±5℃,1±0.5min);组装缓冲液组分为30mm tris-hcl,4mm mgcl2,26μmnad,1mm dtt,50μg/mlbsa;
34、优选地,所述杂交缓冲液组分中peg4000的体积百分比浓度为8%;
35、优选地,向杂交反应体系中加入大肠杆菌dna连接酶或t7 dna连接酶,优选加入大肠杆菌dna连接酶;
36、优选地,在杂交缓冲液中加入相邻的酶切后dna组装序列数为3-12个,取整数;
37、优选地,在杂交缓冲液中加入相邻的酶切后dna组装序列数为6或7个。
38、本发明的有益效果为:
39、本发明提供一种双链dna粘性末端的生成系统在体外大片段基因组dna的合成组装中的用途,该系统可以在双链dna片段两端产生超长和精确的悬臂,悬臂通过可编程dna引导的链切割(dsc)技术产生,可自主设计,没有长度或序列偏好。
40、本发明还提供一种体外迭代组装策略,使用dna引导链切割(dsc)技术处理小dna片段,在小dna片段两端产生超长和精确的悬臂,两端具有超长悬臂的小片段进一步用于体外组装,通过一系列的测试优化含目标悬臂dna片段的组装效率,从而实现体外大片段基因组dna的合成组装。本发明克服了限制性内切酶(res)产生短悬臂的限制并提高大dna分子组装的成功率。本发明以t7噬菌体为例,由pcr扩增子组装成完整的t7噬菌体基因组,并能够产生感染性病毒粒子。最后,利用62个合成dna片段在体外构建了端粒-端粒合成酵母染色体,并将其分离并转移到酵母细胞中。本发明技术通常用于大片段dna分子的无缝构建,且可加速未来的基因组的合成。
- 还没有人留言评论。精彩留言会获得点赞!