含有Salen型四价铂配合物及其制备方法与应用与流程
本发明涉及一种药用化合物及其制备方法与应用,确切讲是一种含有salen型四价铂配合物及其制备方法与应用。
背景技术:
1、恶性肿瘤高发病率和高死亡率给家庭带来了深刻的痛苦和巨大的经济负担。传统的抗肿瘤治疗在抑制肿瘤进展方面作用有限,即使通过手术摘除了肿瘤,也有很多的患者出现局部复发或远处转移。实体瘤的治疗主要为手术治疗,但手术治疗容易导致肿瘤微转移,增加了肿瘤转移的风险。化学疗法可以杀死各种途径转移的癌细胞,从而阻止其转移。铂类抗肿瘤药物在临床上广泛用于恶性肿瘤的治疗。美国fda已批准三种铂类药物上市,包括顺铂、卡铂和奥沙利铂。它们的副作用比较常见的是表现在胃肠道、血液系统、神经系统,以及其他系统或部位。胃肠道不良反应比较常见可能会出现恶心、呕吐、腹泻等相关症状;神经系统的不良反应可能会使患者出现运动或感觉的障碍,或者容易出现耳鸣以及影响听力的状况;血液系统不良反应有可能导致患者出现骨髓抑制的情况,比如可能会出现白细胞、粒细胞、血小板的下降或贫血的状况。铂类抗肿瘤药物是我国肿瘤病人使用最为广泛的化疗药物,但长期使用易产生耐药性、并存在肾毒性、胃肠道不良反应、血液毒性等副作用。参见cn202210295294.x。将二价的铂类药物氧化成四价铂,进一步在结构上引入其他抗肿瘤药效基团是近年来新型铂类抗肿瘤药物的热点研发领域。要想设计出各方面药理指标都超过顺铂的抗肿瘤药物,就必须突破顺铂类药物的限制,寻找一类新的抗肿瘤药物。胰腺癌在所有癌症中更是一种极为凶险的病症,寻找一种对胰腺癌有更好疗效的药物则是业界的一个重大课题。
技术实现思路
1、本发明提供一种可克服现有技术不足的药用化合物。
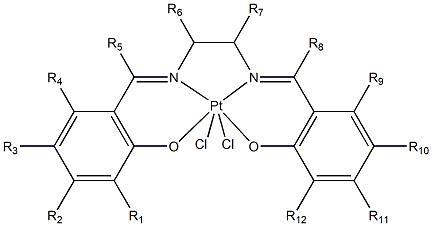
2、本发明的化合物是一种含有salen型四价铂配合物,结构如通式i所示,其中:r1、r2、r3、r4、
3、式1
4、r57、r6、r7、r、r9、r10、r11和r12 分别为h、卤素、氨基、硝基、羧基、羟基、c1~c30的直链或支链烷基、或者为c3~c30的环烷基、或者为c6~c30的芳基中的任一种,且r1、r2、r3、r4、r5、r6、r7、r7、r9、r10、r11和r12相同或不相同。
5、优选地,本发明的含有salen型四价铂配合物为二水杨醛缩乙二胺二氯合铂,其中所有取代基均为h。
6、优选地,本发明的含有salen型四价铂配合物为二(6-甲基水杨醛)缩乙二胺二氯合铂,其中有r1和r12为甲基,其他取代基为h。
7、优选地,本发明的含有salen型四价铂配合物为二(4-羟基水杨醛)缩乙二胺二氯合铂,其中r3和r10为羟基,其他取代基为h。
8、优选地,本发明的含有salen型四价铂配合物为二(4-羧基水杨醛)缩乙二胺二氯合铂,其中r3和r10为羧基,其他取代基为h。
9、优选地,本发明的含有salen型四价铂配合物为二(6-氯水杨醛)缩乙二胺二氯合铂,其中r1和r12为氯,其他取代基为h。
10、优选地,本发明的含有salen型四价铂配合物为6. 二(4-硝基水杨醛)缩乙二胺二氯合铂,其中r3和r10为硝基,其他取代基为h。
11、优选地,本发明的含有salen型四价铂配合物为二(4-叔丁基水杨醛)缩乙二胺二氯合铂,其中r3和r10为叔丁基,其他取代基为h。
12、优选地,本发明的含有为二(4-氨基水杨醛)缩乙二胺二氯合铂,其中r3和r10为氨基,其他取代基为h。
13、前述salen型四价铂配合物的结构如下:
14、。
15、本发明所述配合物的制备方法中合成反应历程如式2示,
16、式2
17、具体的反应为:
18、(1)将化合物a、b、c溶解于乙醇溶液中。a、b、c三者的的摩尔比为1:1:1。在55 ℃水浴条件下,将溶液a和c的混合溶液缓慢滴加到溶液b中,混合溶液继续在55 °c 回流搅拌2 小时。反应完成后收集混合溶液并真空浓缩至后进行柱层析分离,展开剂(二氯甲烷/乙酸乙酯,20:1),收集第二色谱带,旋干溶剂得白色固体,在乙醇/正己烷溶剂中重结晶,得到白色针状晶体化合物d;
19、(2)将摩尔比为1:1的化合物d与ptcl4溶于乙腈溶剂中,室温搅拌反应12~24 h,柱层析纯化制备得到目标化合物——配合物i。
20、优选地,本发明的配合物二水杨醛缩乙二胺二氯合铂制备的反应历程如式3示,
21、式3
22、具体反应为:
23、将2.44 g (20 mmol)水杨醛溶解于50 ml乙醇溶液中,得到溶液a。将0.60 g (10mmol)乙二胺溶解在30 ml乙醇溶液中,得到溶液b。在55 ℃ 水浴条件下,将溶液a缓慢滴加到溶液b中,控制滴速(每秒1-2 滴),4小时滴完后,混合溶液继续在55 °c 回流搅拌2小时。反应完成后收集混合溶液并真空浓缩至5 ml后进行柱层析分离,展开剂(二氯甲烷/乙酸乙酯,20:1),收集第二色谱带,旋干溶剂得白色固体,在乙醇/正己烷溶剂中重结晶,得到白色针状晶体化合物iii,
24、将1.34 g化合物iii(5.0 mmol) 与1.69 g ptcl4 (5.0 mmol) 溶于100 ml 乙腈溶剂中,室温搅拌反应12 h,石油醚柱层析纯化得配合物1。
25、优选地,本发明二(6-甲基水杨醛)缩乙二胺二氯合铂配合物的制备其制备反应历程如式4示:
26、式4
27、具体反应为:
28、将2.72 g (20 mmol)6-甲基水杨醛(化合物iv)溶解于50 ml乙醇溶液中,得到溶液a。将0.60 g (10 mmol)乙二胺溶解在30 ml乙醇溶液中,得到溶液b。在55 ℃ 水浴条件下,将溶液a缓慢滴加到溶液b中,控制滴速(每秒1-2 滴),4小时滴完后,混合溶液继续在55°c 回流搅拌2小时。反应完成后收集混合溶液并真空浓缩至5 ml后进行柱层析分离,展开剂(二氯甲烷/乙酸乙酯,20:1),收集第二色谱带,旋干溶剂得白色固体,在乙醇/正己烷溶剂中重结晶,得到白色针状晶体化合物v,称重为 2.62 g,产率为89%。化合物v的核磁共振数据和高分辨质谱数据如下。1h nmr (dmso-d6): 2.34(6h),3.91 (4h), 5.0 (2h), 6.73(2h), 6.92 (2h), 7.26 (2h) , 8.1 (2h)。hrms:m/z分子离子峰:296.15;
29、将1.48 g化合物v(5.0 mmol) 与1.69 g ptcl4 (5.0 mmol) 溶于100 ml 乙腈溶剂中,室温搅拌反应12 h,石油醚柱层析纯化得配合物2。
30、优选地,本发明的取代基r3和r10分别为羟基,其他取代基为h的铂配合物二(4-羟基水杨醛)缩乙二胺二氯合铂的制备其制备反应历程如式5示,
31、式5
32、具体反应为:取2.76 g (20 mmol)4-羟基水杨醛(化合物vi)溶解于50 ml乙醇溶液中,得到溶液a。将0.60 g (10 mmol)乙二胺溶解在30 ml乙醇溶液中,得到溶液b。在55℃ 水浴条件下,将溶液a缓慢滴加到溶液b中,滴完后混合溶液继续在55 °c 回流搅拌反应完,成后收集混合溶液并真空浓缩至5 ml后进行柱层析分离,展开剂(二氯甲烷/乙酸乙酯,20:1),收集第二色谱带,旋干溶剂得白色固体,在乙醇/正己烷溶剂中重结晶,得到白色针状晶体化合物vii;
33、将1.50 g化合物vii(5.0 mmol) 与1.69 g ptcl4 (5.0 mmol) 溶于100 ml 乙腈溶剂中,室温搅拌反应12 h,石油醚柱层析纯化得配合物3。
34、优选地,本发明的取代基r3和r10分别为羧基,其他取代基为h的二(4-羟基水杨醛)缩乙二胺二氯合铂配合物的制备合成反应历程如式6示,
35、式6
36、具体反应为:将2.32 g (20 mmol)4-羧基水杨醛(化合物viii)溶解于50 ml乙醇溶液中,得到溶液a。将0.60 g (10 mmol)乙二胺溶解在30 ml乙醇溶液中,得到溶液b。在55℃ 水浴条件下,将溶液a缓慢滴加到溶液b中,控制滴速(每秒1-2 滴),4小时滴完后,混合溶液继续在55 °c 回流搅拌2小时。反应完成后收集混合溶液并真空浓缩至5 ml后进行柱层析分离,展开剂(二氯甲烷/乙酸乙酯,20:1),收集第二色谱带,旋干溶剂得白色固体,在乙醇/正己烷溶剂中重结晶,得到白色针状晶体化合物ix;
37、将1.78 g化合物ix(5.0 mmol) 与1.69 g ptcl4 (5.0 mmol) 溶于100 ml 乙腈溶剂中,室温搅拌反应12 h,石油醚柱层析纯化得配合物4,。
38、优选地,本发明配合物二(6-氯水杨醛)缩乙二胺二氯合铂的制备合成反应历程如式7示,
39、式7
40、具体反应为:取将3.13 g (20 mmol)4-羧基水杨醛(化合物x)溶解于50 ml乙醇溶液中,得到溶液a。将0.60 g (10 mmol)乙二胺溶解在30 ml乙醇溶液中,得到溶液b。在55℃ 水浴条件下,将溶液a缓慢滴加到溶液b中,控制滴速(每秒1-2 滴),4小时滴完后,混合溶液继续在55 °c 回流搅拌2小时。反应完成后收集混合溶液并真空浓缩至5 ml后进行柱层析分离,展开剂(二氯甲烷/乙酸乙酯,20:1),收集第二色谱带,旋干溶剂得白色固体,在乙醇/正己烷溶剂中重结晶,得到白色针状晶体化合物xi;
41、将1.69 g化合物xi(5.0 mmol) 与1.69 g ptcl4(5.0 mmol) 溶于100 ml 乙腈溶剂中,室温搅拌反应12 h,石油醚柱层析纯化得配合物4。
42、优选地,本发明配合物二(4-硝基水杨醛)缩乙二胺二氯合铂的制备合成反应历程如式8示,
43、
44、式8
45、具体反应为:取3.66 g (20 mmol)4-硝基水杨醛(化合物xii)溶解于60 ml乙醇溶液中,得到溶液a。将0.60 g (10 mmol)乙二胺溶解在30 ml乙醇溶液中,得到溶液b。在55℃ 水浴条件下,将溶液a缓慢滴加到溶液b中,控制滴速(每秒1-2滴),4小时滴完后,混合溶液继续在55 °c 回流搅拌2小时。反应完成后收集混合溶液并真空浓缩至5 ml后进行柱层析分离,展开剂(二氯甲烷/乙酸乙酯,20:1),收集第二色谱带,旋干溶剂得白色固体,在乙醇/正己烷溶剂中重结晶,得到白色针状晶体化合物xiii,称重为3.52 g;
46、将2.05 g化合物xiii(5.0 mmol) 与1.69 g ptcl4 (5.0 mmol) 溶于120 ml 乙腈溶剂中,室温搅拌反应12 h,石油醚柱层析纯化得配合物6。
47、优选地,本发明配合物二(4-叔丁基水杨醛)缩乙二胺二氯合铂的制备合成反应历程如式9示,
48、式9
49、具体反应为:将3.56 g (20 mmol)4-叔丁基水杨醛(化合物xiv)溶解于60 ml乙醇溶液中,得到溶液a。将0.60 g (10 mmol)乙二胺溶解在30 ml乙醇溶液中,得到溶液b。在55℃ 水浴条件下,将溶液a缓慢滴加到溶液b中,控制滴速(每秒1-2滴),4小时滴完后,混合溶液继续在55 °c 回流搅拌2小时。反应完成后收集混合溶液并真空浓缩至5 ml后进行柱层析分离,展开剂(二氯甲烷/乙酸乙酯,20:1),收集第二色谱带,旋干溶剂得白色固体,在乙醇/正己烷溶剂中重结晶,得到白色针状晶体化合物xv;
50、将1.90 g化合物xv(5.0 mmol) 与1.69 g ptcl4 (5.0 mmol) 溶于120 ml 乙腈溶剂中,室温搅拌反应12 h,石油醚柱层析纯化得配合物7。
51、优选地,本发明二(4-氨基水杨醛)缩乙二胺二氯合铂配合物的制备合成反应历程如式10示,
52、式10
53、具体反应为:将2.74 g (20 mmol)4-氨基水杨醛(化合物xvi)溶解于60 ml乙醇溶液中,得到溶液a。将0.60 g (10 mmol)乙二胺溶解在30 ml乙醇溶液中,得到溶液b。在55℃ 水浴条件下,将溶液a缓慢滴加到溶液b中,控制滴速(每秒1-2滴),4小时滴完后,混合溶液继续在55 °c 回流搅拌2小时。反应完成后收集混合溶液并真空浓缩至5 ml后进行柱层析分离,展开剂(二氯甲烷/乙酸乙酯,20:1),收集第二色谱带,旋干溶剂得白色固体,在乙醇/正己烷溶剂中重结晶,得到白色针状晶体化合物xvii;
54、将1.49 g化合物xvii(5.0 mmol) 与1.69 g ptcl4 (5.0 mmol) 溶于120 ml 乙腈溶剂中,室温搅拌反应12 h,石油醚柱层析纯化得配合物8。
55、本发明的含有salen型四价铂配合物可在制备抗肿瘤药物中的应用,特别是在制备抗胰腺癌症药物中的应用。
56、salen型化合物是一类特殊结构的schiff 碱,由两分子水杨醛与一分子乙二胺缩合而成。salen型化合物与金属配位衍生物的配合物因独特的稳定性、氧化还原性及与dna极好的作用等成为设计以核酸为靶的抗癌药物的一类重要化合物。本发明提供的一类含salen型四价铂配合物,对胰腺癌panc-1/gem产生了较为明显的增殖抑制作用,部分化合物的抗肿瘤活性明显优于顺铂,可作为抗肿瘤的候选药物进行更加深入的研究。如配合物7表现出最优的抗肿瘤活性,对胰腺癌panc-1/gem的半数抑制浓度ic50 最低为1.12 µm。本发明提供的配合物具有全新的骨架结构,具有优异的抗肿瘤活性和较低的细胞毒性,可以进行抗胰腺癌药物开发。
57、本发明具有以下优点和有益效果:
58、针对目前二价铂类抗肿瘤药物在抗肿瘤治疗中存在副作用大和活性低等问题,本发明提供了一类含salen型四价铂配合物,该配合物利用salen型化合物与金属配位衍生物的配合物独特的稳定性、氧化还原性及与dna极好的作用,四价铂高效的抗肿瘤活性,解决二价铂类药物的耐药性,发挥优异的协同抗肿瘤效果。
59、本发明提供的salen型四价铂配合物的合成路线简单,合成原料易得、合成方法容易实现和成本便宜。
- 还没有人留言评论。精彩留言会获得点赞!