一种巨噬细胞焦亡的研究以及抑制方法
本发明涉及化学细胞研究领域,尤其涉及一种巨噬细胞焦亡的研究以及抑制方法。
背景技术:
1、脓毒症是指宿主对感染的免疫应答失调而引起的危及生命的器官功 能障碍。患者常表现为发热、心源性休克、呼吸和/或全身器官功能衰竭。脓毒 症是一个重要的健康问题,也是全球发生危重疾病和死亡的主要原因。仅 2017年,全球就有大约 4890 万新发脓毒症病例,其中与脓毒症相关的死亡人数为 1100 万例,这占全世界死亡总数的 19.7%。
2、尽管近年来医疗技术水平不断提高,脓 毒症依然是造成重症监护室病人死亡的主要原因,长期死亡率为 40%-80%。急 性肺损伤作为脓毒症最常见的并发症,其病理特点是肺泡-毛细血管膜损 伤,中性粒细胞聚集活化,产生大量的炎症和细胞毒性介质,血管通透性增加, 渗出蛋白液,炎症细胞从肺血管向间质和肺泡间隙迁移。人类 ali/ards 发病时的急性病理生理变化包括严重低氧血症,以及肺顺应性降低导致的总胸廓顺应 性下降,而肺水肿的显著增加是由于内皮和上皮细胞的通透性增加,与肺泡上皮 积极清除肺泡液的能力受损有关,这些变化导致严重的通气/灌注不匹配和分流率增加。
3、细胞焦亡是区别于细胞凋亡的新型程序性细胞 死亡,主要由 gasdermin(gsdm)家族蛋白诱导并释放成熟白细胞介素 1-β (interleukin1-β, il-1β)和白细胞介素 18(interleukin18, il-18)产生炎性级联反应。巨噬细胞属免疫细胞,有多种功能,是研究细胞吞噬、细胞免疫和分子免疫学的重要对象。巨噬细胞容易获得,便于培养,并可进行纯化。巨噬细胞属不繁殖细胞群,在条件适宜下可生活2~3周,多用做原代培养,难以长期生存。
4、目前对于巨噬细胞焦亡的研究并不是很顺利,难以发现其焦亡的过程与何种信息有关。
技术实现思路
1、本发明的目的是为了解决现有技术中存在的缺点,而提出的一种巨噬细胞焦亡的研究以及抑制方法。
2、为了实现上述目的,本发明采用了如下技术方案:一种巨噬细胞焦亡的研究以及抑制方法,所述抑制方法包括:atp合酶抑制因子1的研究,线粒体atpif1蛋白的酵解以及atpif1在ali小鼠心肌组织和lps诱导的巨噬细胞中的表达;
3、线粒体atpif1蛋白的酵解过程为我们揭示了其能量调控的潜在机制;在酵解过程中,atpif1通过其特定的结构域与atp合酶相互作用,影响atp的合成和分解,进而调控细胞的能量状态;
4、在ali小鼠的心肌组织中,atpif1的表达量明显升高;这可能是由于心肌细胞在应对缺氧、炎症等刺激时,需要更多的能量来维持其正常功能;而atpif1通过调控atp合酶的活性,影响atp的合成和分解,从而满足心肌细胞对能量的需求;
5、在lps诱导的巨噬细胞中,atpif1的表达量则呈现下降趋势;lps作为一种细菌内毒素,能够激活巨噬细胞并诱导其产生一系列炎症反应;在这个过程中,巨噬细胞需要大量的能量来支持其免疫反应;然而,atpif1的表达降低可能导致atp合酶的活性下降,从而影响atp的合成和分解,使巨噬细胞在能量供应上受到限制。
6、作为上述技术方案的进一步描述:
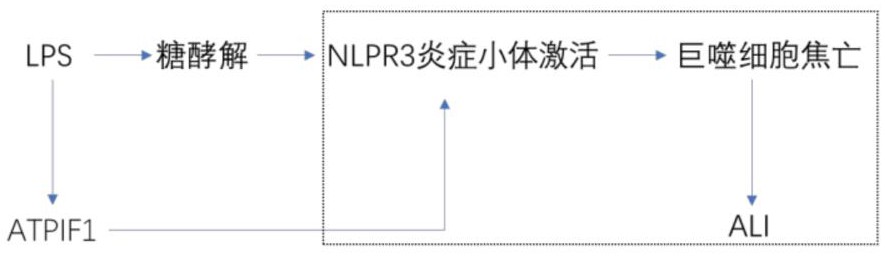
7、巨噬细胞的焦亡:内毒素可通过经典途径激活 toll 样受体 4(toll-likereceptors 4,tlr4)激活 nlrp3 炎症小体,促进半胱氨酸蛋白酶 1(caspase 1)剪切活化并进一步剪切 gasdermind(gsdmd)形成活性 gsdmd-n 末端及剪切 pro-il-1β 和 pro-il-18 促进其成熟,在细胞膜上形成膜孔释放成熟 il-1β和 il-18 诱发炎性级联反应,促进肺泡巨噬细胞焦亡,最终加重 ali/ards,而抑制 nlrp3 炎症小体的激活则可以阻断巨噬细胞焦亡减轻 ali/ards。
8、具体的、内毒素,作为一种强烈的炎症刺激因子,通过其独特的信号传导机制,在肺部炎症疾病中发挥着关键作用。一旦内毒素通过经典途径激活了toll样受体4(tlr4),一系列复杂的细胞反应便随之触发。其中,nlrp3炎症小体的激活是这一系列反应中的关键环节。
9、nlrp3炎症小体是一个多蛋白复合物,其激活需要多种信号的共同作用。在内毒素的刺激下,nlrp3炎症小体被激活,进而促进了半胱氨酸蛋白酶1(caspase 1)的剪切活化。这一活化的caspase 1进而剪切gasdermind(gsdmd),使其形成具有生物活性的gsdmd-n末端。
10、与此同时,活化的caspase 1还剪切了pro-il-1β和pro-il-18,使其转化为成熟的il-1β和il-18。这两种成熟的炎症因子随后在细胞膜上形成的膜孔中释放,从而触发了广泛的炎性级联反应。这种级联反应不仅加剧了肺部的炎症反应,还促进了肺泡巨噬细胞的焦亡。
11、肺泡巨噬细胞的焦亡是ali/ards疾病进程中的重要事件。这些细胞在受到强烈刺激时,会经历一种程序性细胞死亡,即焦亡。焦亡的巨噬细胞会释放大量的炎症介质和细胞碎片,进一步加剧了肺部的损伤和炎症反应。
12、因此,抑制nlrp3炎症小体的激活成为了阻断巨噬细胞焦亡、减轻ali/ards的有效策略。通过药物或其他手段抑制nlrp3的激活,可以阻断其下游的炎症反应链,从而减轻肺部的损伤和炎症反应。这为ali/ards的治疗提供了新的思路和方法。
13、作为上述技术方案的进一步描述:
14、atp合酶抑制因子1具有重要的免疫调控功能,其敲除可减轻 lps 诱导的巨噬细胞焦亡和ali。
15、进一步的研究发现,atp合酶抑制因子1的敲除不仅影响巨噬细胞的焦亡过程,还可能对免疫细胞的增殖、分化和凋亡等过程产生深远影响。在敲除atp合酶抑制因子1的个体中,免疫细胞对于一些病原体的应答可能会发生改变,这可能有助于我们更深入地理解免疫系统的工作机制。
16、同时,atp合酶抑制因子1也可能与其他免疫分子存在相互作用,共同调控免疫应答。这些相互作用可能涉及到信号通路的激活或抑制,以及免疫细胞之间的通讯等。因此,对atp合酶抑制因子1的深入研究,有望揭示更多关于免疫调控的新机制和新靶点。
17、作为上述技术方案的进一步描述:
18、线粒体 atpif1 蛋白可能通过介导糖酵解加重巨噬细胞焦亡及 ali。
19、进一步的研究表明,线粒体atpif1蛋白在巨噬细胞焦亡和急性肺损伤(ali)的过程中起到了关键的调控作用。这一蛋白通过介导糖酵解过程,可能加剧了巨噬细胞在炎症环境下的损伤反应。
20、巨噬细胞作为免疫系统的关键组成部分,在抵御病原体和调节炎症反应方面发挥着至关重要的作用。然而,在炎症过程中,巨噬细胞可能遭受过度激活,导致细胞焦亡,进而引发组织损伤。线粒体atpif1蛋白的参与,使得这一过程变得更为复杂。
21、线粒体atpif1蛋白通过介导糖酵解过程,可能加剧了巨噬细胞在炎症环境下的焦亡反应,进而促进了ali的发生和发展。这一发现为我们深入理解巨噬细胞焦亡和ali的机制提供了新的视角,也为未来开发针对这些疾病的治疗策略提供了新的思路。
22、作为上述技术方案的进一步描述:
23、线粒体atp合成酶抑制因子 1(atp synthase inhibitory factor 1, atpif1)是由核编码的 12kda 蛋白质分子,在所有组织中均广泛表达,尤其是能量需求高的组织和器官,主要通过与线粒体f0f1-atp合成酶(或称为线粒体呼吸链复合体v)相互作用调控atp的合成和水解,从而调节细胞代谢等生物学功能;
24、线粒体f0f1-at合成酶是由线粒体基质中的可溶性催化 f1 区和膜包埋的 f0 区组成。
25、作为上述技术方案的进一步描述:
26、atpif1在lps诱导的小鼠肺组织及巨噬细胞中显著增加,atpif1 的特异性敲除可减轻 lps 诱导的ali;并且在lps诱导的ali中与炎症相关的基因大量富集,尤其是与巨噬细胞焦亡相关的炎症基因,atpif1flox小鼠在 lps 刺激后 il-1β、il-18和nlrp3显著增加,而atpif1特异性敲除后 il-1β、il-18和nlrp3明显减少。
27、作为上述技术方案的进一步描述:
28、smad4 可作用于糖酵解酶pkm2和线粒体atpif1,增加足细胞中糖酵解和减少氧化磷酸化atpif1可抑制f0f1-atp合成酶活性,阻断线粒体呼吸电子传递链中电子流动,促使线粒体 ros 产生并稳定hif-1α,最终激活糖酵解,而atpif1特异性敲除后可阻断该代谢重编程。
29、作为上述技术方案的进一步描述:
30、抑制方法的步骤包括:
31、atpif1 在 ali 小鼠心肌组织和 lps 诱导的巨噬细胞中的表达;
32、s1、建立ali模型:在lps 诱导的ali模型中,随lps刺激时间的逐渐增加,atpif1的蛋白和mrna 表达出现先升高后降低的趋势,且均高于基线水平;(与0 h 相比,*p<0.05);
33、s2、atpif1在lps诱导的thp-1巨噬细胞中的蛋白与mrna表达情况。免疫印迹western blot和rt-pcr结果显示:lps刺激巨噬细胞后,atpif1蛋白表达在lps刺激后的第6h开始出 现升高,atpif1 mrna表达则在lps 刺激后持续升高;(与0 h相比,*p<0.05);
34、s3、atpif1在lps诱导的beas-2b上皮细胞和huvec内皮细胞中的蛋白表达情况;免疫印迹western blot和rt-pcr结果显示:lps刺激beas-2b上皮细胞和huvec内皮细胞后,atpif1蛋白表达基本不变,无显著性差异;
35、s4、生物信息学技术分析与atpif1基因启动子区域相结合的转录因子。生物信息学技术分析结果显示在atpif1基因启动子区域共检测到46个调控转录因子,经筛选后atf6和klf4两个转录因子与atpif1转录强相关;
36、s5、atf6和klf4对atpif1的转录调控作用;免疫印迹western blot结果显示:用atf6的sirna抑制其功能后,pbs组和lps组atpif1的表达均被显著抑制;而用klf4的sirna抑制其功能后,lps组atpif1的表达较pbs组显著增加;提示转录因子atf6可参与调控atpif1的转录,而klf4不可以。
37、作为上述技术方案的进一步描述:
38、抑制方法的步骤包括:
39、atpif1敲除改善小鼠脓毒症肺损伤;
40、s1、验证atpif1特异性基因敲除小鼠的敲除效果。免疫印迹western blot结果显示:atpif1mko小鼠atpif1蛋白无表达,说明atpif1基因敲除小鼠模型构建成功;
41、s2、lps注射12h后,各组小鼠肺损伤、肺部炎症水平的比较;h&e染色结果显示atpif1敲除可明显抑制ali小鼠肺组织中炎症细胞浸润,减轻ali。相关试剂盒检测结果显示atpif1敲除可抑制肺组织中mpo的活性、减少肺泡灌洗液中总细胞数和总蛋白量;(与control+atpif1 flox,*p<0.05;与lps+atpif1 flox,#p<0.05);
42、s3、lps注射后各组小鼠的生存曲线。atpif1敲除可显著延长ali小鼠的生存时间;(与lps+ atpif1 flox,#p<0.05)。
43、作为上述技术方案的进一步描述:
44、抑制方法的步骤包括:
45、atpif1敲除减轻脓毒血症小鼠巨噬细胞焦亡;
46、s1、与atpif1相关性强的基因主要为炎症相关基因,尤其是与细胞焦亡相关的基因;
47、s2、c lps 注射12 h 后,各组小鼠肺泡灌洗液中细胞因子的表达。elisa分析结果显示:atrpif1敲除可明显抑制ali小鼠肺泡灌洗液中细胞因子il-1β和il-18的升高;(与control+atpif1 flox,*p<0.05;与lps+atpif1 flox,#p<0.05);
48、s3、glps 注射12 h 后,各组小鼠肺组织巨噬细胞焦亡标志物蛋白表达。免疫印迹western blot 结果显示:atrpif1敲除可明显抑制ali小鼠肺组织中nlrp3、il-1β和il-18的升高,表明atrpif1敲除具有抑制ali小鼠肺巨噬细胞焦亡的效应;(与control+atpif1flox,*p<0.05;与lps+atpif1 flox,#p<0.05)。
49、本发明具有如下有益效果:
50、本发明进一步提供了atpif1在脓毒症肺损伤中的具体作用机制。
51、在深入研究过程中,我们发现atpif1在脓毒症肺损伤中起到了关键作用。脓毒症肺损伤发生时,lps的刺激导致atpif1的表达增加,进而促进了il-1β、il-18和nlrp3等炎症因子的显著上升。然而,当atpif1被特异性敲除后,这些炎症因子的表达明显减少,表明atpif1在调控脓毒症肺损伤中的炎症反应起到了重要作用。
52、进一步的研究揭示了atpif1的作用机制。atpif1通过抑制f0f1-atp合成酶的活性,阻断线粒体呼吸电子传递链中的电子流动。这一过程导致线粒体ros的产生增加,进而稳定了hif-1α,并激活了糖酵解过程。这种代谢重编程是脓毒症肺损伤中的一个重要环节,而atpif1的敲除能够阻断这一过程,从而减轻肺损伤。
53、此外,我们还发现atpif1在ali小鼠心肌组织和lps诱导的巨噬细胞中的表达存在特定的变化模式。在ali模型中,随着lps刺激时间的增加,atpif1的表达呈现出先升高后降低的趋势。而在lps诱导的巨噬细胞中,atpif1的蛋白和mrna表达也呈现出明显的变化。这些发现为我们进一步理解atpif1在脓毒症肺损伤中的作用提供了重要线索。
54、综上所述,本发明揭示了atpif1在脓毒症肺损伤中的关键作用及其作用机制,为脓毒症肺损伤的治疗提供了新的思路和方法。通过特异性敲除atpif1,我们可以有效地抑制脓毒症肺损伤中的炎症反应和代谢重编程过程,从而减轻肺损伤并提高患者的生存率。这一发现为未来的药物研发提供了新的靶点,有望为脓毒症肺损伤的治疗带来突破性的进展。
- 还没有人留言评论。精彩留言会获得点赞!