一种美洛昔康降解杂质的制备方法与流程
本发明属于药物合成,具体涉及一种美洛昔康降解杂质的制备方法。
背景技术:
1、美洛昔康是一种环氧合酶-2(cyclooxygenase-2,cox-2)抑制剂,属于非甾体抗炎药(nsaids),其作用机制是通过抑制环氧合酶(cyclooxygenase,cox)来阻止致炎的前列腺素合成。美洛昔康与传统的nsaids相比,抗炎镇痛效果相似,且胃肠道耐受性显著提高。美洛昔康的cox-2/cox-1值大大低于其他临床上常用的nsaids,因此在临床上美洛昔康的应用更为广泛。
2、为了保证用药安全,原料药/制剂中的每一个杂质都必须进行安全性评估也就是说必须建立保证安全性的杂质限度。《中国药典》对美洛昔康杂质的总量进行了限定:供试品溶液色谱图中如有杂质峰,单个杂质峰面积不得大于对照溶液主峰面积的0.5倍(0.5%),各杂质峰面积的和不得大于对照溶液主峰面积(1.0%)。而ich准则要求:药物中杂质的限度为0.1%(对毒性药物限度更低),高于此水平的所有未知杂质应鉴别出来,而更重要的是,所有高于0.15%的杂质应研究其毒性。
3、
4、目前,针对美洛昔康降解杂质的研究较少,研究发现在美洛昔康的稳定性实验考察期间,会降解产生如式1结构所示的杂质化合物,且该杂质随着稳定性考察时间的延长,杂质含量逐渐增大,容易超过ich指导原则规定的界定限度(0.15%),对药品的安全性、有效性和质量可控性有重要的影响,因此本领域有必要提供一种美洛昔康降解杂质的制备方法。
技术实现思路
1、本发明针对现有技术存在的问题,提供了一种美洛昔康降解杂质的制备方法。
2、为实现上述目的,本发明采用的技术方案如下:
3、一种美洛昔康降解杂质的制备方法,包括如下步骤:
4、(1)将美洛昔康溶于有机溶剂,降解,得降解物;
5、(2)对降解物进行柱色谱分离,纯化,即得,
6、其中,步骤(1)中所述降解为光照降解、高温降解或氧化降解,
7、所述美洛昔康降解杂质的结构式如式1所示:
8、
9、优选地,所述光照降解的光强为4000-8000lx,光照降解的时间为5-11天,光源总照度不低于1.2×106lux·hr,近紫外灯能量不低于200w·hr/m2。
10、更优选地,所述光照降解的光强为5500-6500lx,光照降解的时间为8-10天,光源总照度为1.3×106lux·hr,近紫外灯能量为250w·hr/m2。
11、进一步优选地,所述光照降解的光强为6000lx,光照降解的时间为9天。
12、优选地,步骤(1)中所述有机溶剂选自二甲基甲酰胺、二甲基乙酰胺、二甲亚砜、丙酮、异丙醇和六氟异丙醇中的一种或多种。
13、更优选地,所述有机溶剂为二甲亚砜、异丙醇或六氟异丙醇。
14、优选地,所述美洛昔康溶于有机溶剂之后的浓度为0.1-10mg/ml。
15、更优选地,所述美洛昔康溶于有机溶剂之后的浓度为5-10mg/ml。
16、在美洛昔康溶解过程中,可通过强力振摇、涡旋或超声等方式助溶。
17、优选地,所述高温降解的温度为40-150℃,高温降解的时间为5-12h。
18、更优选地,所述高温降解的温度为130-140℃,高温降解的时间为9-11h。
19、进一步优选地,所述高温降解的温度为135℃,高温降解的时间为10h。
20、所述氧化降解的过程包括与体积浓度为3-10%的双氧水混匀,所述氧化降解的温度为30-50℃,氧化降解的时间为3-8h。
21、优选地,所述氧化降解的过程包括与体积浓度为6-10%的双氧水混匀,所述氧化降解的温度为40-50℃,氧化降解的时间为4-6h。
22、更优选地,所述氧化降解的过程包括与体积浓度为10%的双氧水混匀,所述氧化降解的温度为50℃,氧化降解的时间为5h。
23、通过如下色谱参数进行杂质的分离,最大限度的减少了其它降解组分对目标物的干扰,得到的杂质更纯净。
24、优选地,步骤(2)中所述柱色谱分离的条件包括:流动相a为体积比为70:30-95:5的磷酸盐缓冲液与甲醇的混合物,流动相b为体积比为6:4-8:2的甲醇与乙腈的混合物,所述柱色谱分离的洗脱方式为梯度洗脱。
25、更优选地,流动相a为体积比为90:10的磷酸盐缓冲液与甲醇的混合物,流动相b为体积比为7:3的甲醇与乙腈的混合物,
26、优选地,所述磷酸盐缓冲液为磷酸二氢钾溶液、磷酸氢二钾溶液、磷酸二氢钠溶液、磷酸氢二钠溶液、磷酸二氢铵溶液或磷酸氢二铵溶液,所述磷酸盐缓冲液的ph为4.5-6.5。
27、更优选地,所述磷酸盐缓冲液为磷酸氢二铵溶液,所述磷酸盐缓冲液的ph为5.5。
28、优选地,所述梯度洗脱的洗脱程序为:
29、0-10min,a:b为100-80:0-20,v/v;
30、15-25min,a:b为80-60:20-40,v/v;
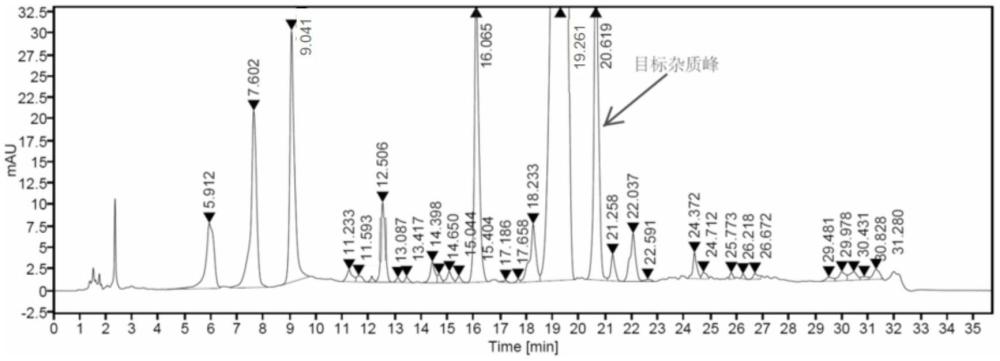
31、25.1-35min,a:b为100-80:0-20,v/v。
32、更优选地,所述梯度洗脱的洗脱程序为:
33、0-10min,a:b为95:5,v/v;
34、15-25min,a:b为70:30,v/v;
35、25.1-35min,a:b为95:5,v/v。
36、优选地,步骤(2)中所述柱色谱分离的条件还包括:色谱柱为shim-pack gis c18或usha c18,检测波长为250-270nm,流速为1-20ml/min,柱温为25-45℃。
37、更优选地,所述色谱柱为shim-pack gis c18,检测波长为260nm,流速为10ml/min,柱温为30℃。
38、优选地,步骤(2)中所述柱色谱分离结束后还包括:收集相对保留时间为1.05-1.10的液体组分。
39、接取的杂质流出组分液通过以下方式进行进一步纯化干燥,该纯化干燥方式在药品研发实验室更容易实现,纯化干燥效率更高,且更能保护分离纯化出的杂质质量。
40、优选地,所述纯化包括将液体组分溶于非极性溶剂,萃取,挥干非极性溶剂,干燥,即得。
41、优选地,所述非极性溶剂为乙酸乙酯、甲苯、正己烷、二氯甲烷、三氯甲烷或正庚烷。
42、更优选地,所述非极性溶剂为乙酸乙酯、甲苯或正庚烷。
43、优选地,所述挥干非极性溶剂的方式为氮气吹扫或旋蒸,所述氮气吹扫的气体流量为1-20lpm,氮气吹扫的温度为25-60℃;所述旋蒸的转速为20-180rpm,旋蒸的温度35-60℃,旋蒸的真空度为250-500mbar。
44、更优选地,所述氮气吹扫的气体流量为8-12lpm,氮气吹扫的温度为35-45℃。
45、更优选地,所述旋蒸的转速为40-180rpm,旋蒸的温度40-50℃,旋蒸的真空度为300-450mbar。
46、优选地,所述干燥为减压干燥,所述干燥的温度为45-80℃,干燥的压力小于2.67kpa,干燥的时间为5-24h。
47、优选地,所述干燥需使用干燥剂,所述干燥剂为五氧化二磷、无水氯化钙或硅胶。
48、本发明还提供了上述制备方法制备得到的美洛昔康降解杂质在作为美洛昔康杂质对照品中的应用。
49、本发明还提供了上述制备方法制备得到的美洛昔康降解杂质的纯度检测方法,包括将上述制备方法纯化得到的美洛昔康降解杂质与稀释剂混合,进行液相色谱检测,即得。
50、优选地,所述液相色谱的条件包括:色谱柱为welch ultimate xb-c18(150mm×4.6mm、5μm)或ymc-triart-c18(150mm×4.6mm、3μm);流动相a为浓度为20-40mmol/l的磷酸二氢钾溶液或乙酸铵溶液,流动相b为乙腈;检测波长为250-270nm;流速为0.8-1.2ml/min;柱温为25-45℃。
51、优选地,流动相a为浓度为30mmol/l的磷酸二氢钾溶液或乙酸铵溶液,检测波长为260nm,流速为1ml/min,柱温为30℃。
52、优选地,所述稀释剂为体积比为2:8-8:2的乙醇与异丙醇的混合物,所述美洛昔康降解杂质在稀释剂中的质量浓度为0.5-5mg/ml。
53、优选地,所述液相色谱的洗脱方式为梯度洗脱,所述梯度洗脱的洗脱程序为:
54、0-15min,a:b为90-70:10-30,v/v;
55、15-25min,a:b为70-90:30-10,v/v;
56、25-35.1min,a:b为90-70:10-30,v/v;
57、35.1-40min,a:b为90-70:10-30,v/v。
58、相对于现有技术,本发明具有以下有益效果:
59、(1)本发明提供了美洛昔康降解杂质的制备方法,通过制备液相分离条件和纯化方法对降解物进行精制,能够高效得到纯度不低于98.0%的美洛昔康降解杂质,可在原料药和制剂生产的质量标准研究和质量控制中作为对照品,为美洛昔康的质量研究、标准研究、稳定性研究和药品不良反应的机制研究提供了基础。
60、(2)本发明还提供了美洛昔康降解杂质的纯度检测方法,该检测方法操作简便,结果准确,能快速准确的测定美洛昔康降解杂质的纯度。
- 还没有人留言评论。精彩留言会获得点赞!