一种检测结核分枝杆菌复合群耐药基因试剂盒及耐药性检测方法
本发明属于分子生物学检测,特别是关于细菌耐药性的诊断技术。更具体地,本发明提供了一种利用靶向纳米孔测序技术对结核分枝杆菌复合群耐药基因进行精确、快速测序并实施耐药性预测的方法。此方法能够有效地识别多种耐药基因,为临床治疗提供重要的耐药信息,从而促进个性化医疗和精准治疗策略的制定。
背景技术:
1、结核病(tuberculosis)在全球公共卫生领域中仍然是一个重大挑战。根据2022年的统计数据,全球结核病患者达到1060万例,其中新增确诊病例高达750万例。结核分枝杆菌复合群(mycobacterium tuberculosis complex,mtbc)的耐药性使得疾病的控制和治疗更加复杂。目前,全球大约有410,000人患有耐药性结核病,但只有约40%的病例得到了准确的诊断和适当的治疗。
2、传统的基于培养的表型药物敏感性试验(pdst)虽能提供药敏诊断信息,但耗时长且操作复杂。分子诊断技术,如cepheid xpert mtb/rif和ultra检测,能快速提供灵敏的诊断结果,尤其适用于疑似肺结核病例。然而,这些方法只能检测有限的耐药突变,可能遗漏其他关键的基因组靶点。因此,迫切需要一种综合检测方法,能全面评估药物耐药性特征,确保有效治疗。靶向下一代测序技术被世界卫生组织(who)认为是管理耐药结核病的关键技术,它能够迅速提供全面的药物敏感性信息,极大地加快临床决策的过程。基于二代测序平台的短扩增子靶向测序方法由于受到测序读长的限制,可能导致关键变异的漏检,并且其在引物设计和数据处理上复杂度较高,增加了方法的操作难度。相比之下,采用长扩增子的靶向测序技术则基于长读长测序平台,如纳米孔测序平台,能够覆盖更长的基因片段,简化了引物设计和数据处理流程,并且能够提供更全面的遗传信息。此外,其文库构建和测序操作流程也更加简洁,有效减少了实验的整体复杂性。然而,纳米孔测序平台原始测序数据的准确度较低,为确保检测的可靠性,通常需要更深的测序深度,这可能会增加检测的时间和成本。此外,该平台还面临着缺乏成熟、一体化数据分析流程的挑战,这对于快速且准确地识别耐药性基因变异尤其关键。
3、本发明涉及一种基于纳米孔测序平台的长扩增子靶向测序方法,旨在通过多重pcr技术直接从临床样本中扩增结核分枝杆菌复合群的近20个关键耐药基因,并预测其对16种常用抗结核药物的耐药性。本发明还包括开发一套一体化数据分析流程,能自动从原始测序数据中提取信息并生成详尽的耐药性预测报告。通过优化关键参数,本发明提高了检测的准确度,并降低了获得准确结果所需的测序深度,有效缩短了检测时间,降低了检测成本。本发明的技术方案优化了实验和数据分析流程,增强了方法的可操作性和效率,为制定更精确的治疗策略提供了技术支撑。本发明在结核分枝杆菌复合群耐药性检测领域具有广泛的应用前景,有望成为耐药结核病实验室检测体系的重要组成部分。此外,本发明的成本效益比和实用性将对公共卫生领域产生显著影响。
技术实现思路
1、本发明旨在提供一种结核分枝杆菌复合群耐药性检测方法。
2、本发明涉及一种基于纳米孔测序平台的长扩增子靶向测序方法,使用多重pcr技术直接从临床样本中扩增结核分枝杆菌复合群的关键耐药基因,检测样本对16种常用抗结核药物的耐药性。
3、本技术方案旨在通过简化实验流程和优化数据分析过程,提高检测的准确性和效率,从而加快临床决策的过程。首先,本发明的靶向测序方法可以直接应用于临床样本,无需对结核分枝杆菌复合群进行分离培养,大幅节省了时间和成本。此外,一次实验即可同时测序约20个与耐药性相关的基因,显著提高了实验效率。整个实验流程简单快速,具有很强的适用性。其次,本发明还包括一套一体化数据分析流程,该流程利用先进的生物信息学工具从原始测序数据中自动提取关键信息,并生成详尽的耐药性预测报告。通过参数优化,如读长覆盖、质量分数阈值和变异频率阈值,确保即便在较低的测序深度下也能达到高准确性的结果,显著提高变异检测的灵敏度和特异性。通过这些技术优化,本发明不仅弥补了现有技术的局限,还有助于深入研究结核分枝杆菌复合群的耐药进化规律和发现新的变异。这一全新的方法在结核分枝杆菌复合群耐药性检测领域具有广泛的应用前景,有望成为耐药结核病实验室检测体系的重要组成部分。本技术的应用能够为制定个性化和精准的治疗方案提供及时且准确的科学依据,推动精准医疗和个性化治疗策略的发展。
4、本发明为此提供一种用于测序结核分枝杆菌复合群耐药相关基因的试剂盒。
5、本发明所述试剂盒中包括seq id no.1-seq id no.40的多核苷酸。
6、其中,seq id no.1-seq id no.40列表如下:
7、
8、
9、根据需要,本发明的试剂盒中还可以包括有利于实验室操作的试剂,如:溶剂,缓冲溶剂,辅助材料等。
10、本发明的试剂盒,可以包括以上组分制备而成的试剂,试剂的配制方法均为常规技术,只需要将各种原材料在常温下混合均匀即可,无需特殊设备和条件。
11、本发明的试剂盒,可以将不同试剂分别盛装,再一同包装在同一包装盒内,使用时根据说明书中描述的方法进行操作。
12、本发明进一步提供本发明试剂盒的使用方法以及耐药基因的检测方法,所述方法,包括以下步骤:
13、步骤1,样本的获得:
14、主要采集患者的痰液作为检测样本。
15、步骤2,dna提取:
16、采用dna提取技术提取样本中的dna。
17、步骤3,多重pcr扩增:
18、采用多重pcr扩增得到pcr产物(扩增子)。
19、步骤4,oxford nanopore technologies(ont)的gridon纳米孔测序仪测序:
20、采用ont的gridon纳米孔测序仪对pcr产物(扩增子)进行测序。
21、步骤5,数据分析:根据测序结果,可以得知样本中是否含有耐药相关突变,
22、从而判断样本对哪些药物耐药。
23、其中,所述耐药基因,列表如下:
24、表1
25、 基因名称 与结核分枝杆菌复合群耐药相关性 rpob 与利福平耐药相关 emba 与乙胺丁醇耐药相关 embb 与乙胺丁醇耐药相关 pnca 与吡嗪酰胺耐药相关 rrs 与链霉素、卷曲霉素、卡那霉素、阿米卡星耐药相关 gid 与链霉素耐药相关 rpsl 与链霉素耐药相关 katg 与异烟肼耐药相关 ahpc 与异烟肼耐药相关 inha 与异烟肼、乙硫异烟胺、丙硫异烟胺耐药相关 etha 与乙硫异烟胺、丙硫异烟胺耐药相关 gyra 与左氧氟沙星、莫西沙星耐药相关 gyrb 与左氧氟沙星、莫西沙星耐药相关 rrl 与利奈唑胺耐药相关 rplc 与利奈唑胺耐药相关 ddn 与德拉马尼耐药相关 tlya 与卷曲霉素耐药相关 eis 与卡那霉素、阿米卡星耐药相关 rv0678 与贝达奎林、氯法齐明耐药相关
26、其中,所述耐药,是指对以下药物耐药:
27、利福平,乙胺丁醇,吡嗪酰胺,链霉素,卷曲霉素,卡那霉素,阿米卡星,异烟肼,乙硫异烟胺,丙硫异烟胺,左氧氟沙星,莫西沙星,利奈唑胺,德拉马尼,贝达奎林,氯法齐明。
28、其中,耐药基因包括:emba,rpsl,ahpc,gid,embb,katg,inha,pnca,rrs,rpob,gyrb,eis,rv0678,gyra,rplc,ddn,tlya,rrl,etha,菌种鉴定基因为hsp65。
29、本发明步骤3中所述多重pcr扩增,需要使用本发明特别设计的pcr引物,19个耐药相关基因与1个菌种鉴定基因的pcr引物分为正向引物和反向引物,每一种耐药基因的引物列表如下:
30、表2
31、
32、优选的,本发明的检测方法,包括以下步骤:
33、步骤1,样本的获得:
34、主要采集患者的痰液作为检测样本。
35、步骤2,dna提取:
36、采用dna提取技术提取样本中的dna。
37、步骤3,多重pcr扩增:
38、1)多重pcr反应体系:
39、该方法可直接应用于临床痰液样本dna,使用的pcr体系和反应条件见表3和表4。
40、表3:多重pcr反应体系
41、 成分 添加体积 kappa ready mix 12.5μl 引物池 2μl dna模板 可变 无核酸酶水 可变 总体积 25μl
42、表4:多重pcr反应条件
43、 名称 反应条件 预变性 95℃300s 变性 98℃20s 退火 65℃15s 延伸 72℃105s 循环数 35 补充延伸 72℃105s
44、2)引物池中各引物对浓度:
45、通过多次预实验,我们对引物池中扩增不同基因的引物对加入量进行了优化(表5),经过优化,各个基因的pcr扩增效率能够达到基本一致。
46、表5:引物池组成
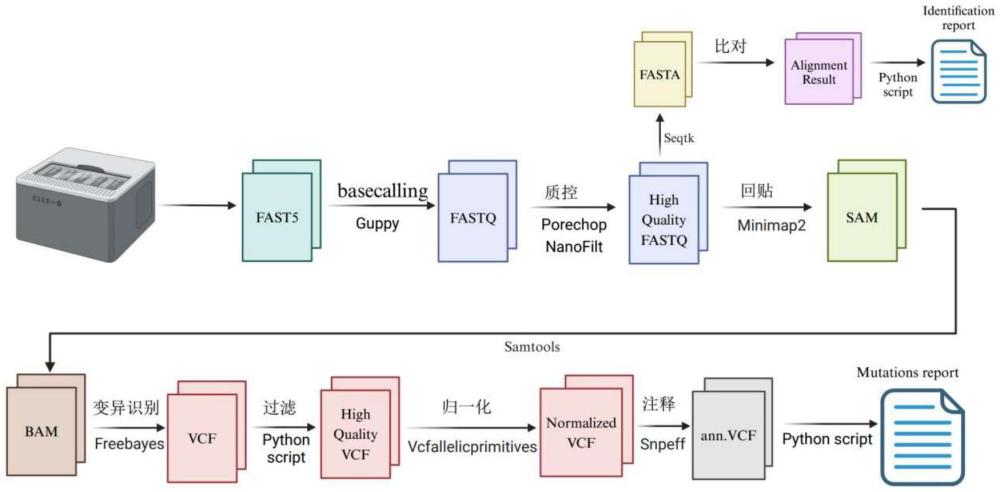
47、
48、
49、
50、步骤4,纳米孔gridon测序仪测序:
51、在多重pcr反应完成后,首先对产物进行核酸定量。每个样本的两个pcr产物管中的产物被设计为不同的基因集,以确保检测的全面性。因此,对这两个管中的产物进行等量混合是必要的,这样操作能确保每个样本的测序文库包含所有目标基因。随后,根据定量结果,为每个样本准备等量的混合pcr产物用于文库构建。在文库构建阶段,首先为每个样本的pcr产物连接带有条形码的适配片段,这对于后续能够在生物信息学流程中准确区分和追踪每个样本至关重要。条形码适配片段连接完成后,将所有样本的pcr产物进行适当的混合,并通过磁珠纯化技术进行处理,完成测序文库的构建。最终,将构建好的文库加载至测序芯片,并在纳米孔gridion测序仪上启动测序。这个过程确保了从每个样本中获得全面的基因覆盖,为后续的分析和解读提供了全面且详尽的遗传信息。
52、步骤5,数据分析:
53、首先,根据参与测序的样本数量,我们确保获得足够的原始数据量,以支撑深入且全面的分析。随后,按照图1所示的分析流程,使用特定的生物信息学软件对原始测序数据进行初步处理。这一阶段的处理包括数据质量评估、读取数据的校正以及必要的数据过滤,以消除技术误差和偏差。在数据处理后,进入详细的生物信息学分析阶段,其中包括对比对后数据的进一步分析,以识别和验证耐药相关的遗传变异。在这一过程中,特别关注样本间的差异性以及任何可能影响数据解读的质量问题。通过采用适当的统计和计算方法,对疑似的数据异常进行校正处理,确保分析结果的高准确性和高可靠性。最后,整合所有分析数据,生成详细的耐药性检测报告。此报告将为临床决策提供科学依据,帮助医生选择最合适的治疗方案。整个分析流程旨在通过高标准的数据处理和精确的遗传分析,提供最准确的耐药性评估。
54、本发明的核心在于,本发明提供了全新引物,所述引物的设计方法如下:
55、本发明的引物设计方法依据特定目标基因序列特征,采用生物信息学工具和软件进行引物设计。首先,通过分析目标基因的保守区域与变异区域,选择适合进行扩增的序列作为引物结合位点。接着,使用如primer3等软件计算引物的理想长度、tm(熔解温度)、gc含量以及是否存在二聚体和发夹结构等可能影响pcr反应的因素。之后,进行引物特异性验证,确保所设计的引物不会与非目标基因序列产生非特异性结合。最终,通过实验验证引物的扩增效率和特异性,确保其在实际应用中的有效性和准确性。
56、此外,本发明的引物设计还考虑了以下关键参数的优化:
57、1.序列特异性:确保引物仅与目标序列特异性结合,避免交叉反应。
58、2.扩增效率:通过优化引物的长度和gc含量,提高pcr扩增效率。
59、3.熔解温度匹配:设计引物对的熔解温度尽可能匹配,以优化pcr反应的温度循环条件。
60、本发明所述检测方法包括以下重要名词术语:
61、pcr反应:聚合酶链式反应,是一种体外扩增特定dna片段的分子生物学技术。通过温度循环,利用dna聚合酶复制目标dna序列。
62、文库构建:将dna或rna样本制备成可以进行测序的分子集合的过程。包括dna片段化、末端修复、接头连接等步骤,为后续测序做准备。
63、测序反应:测序反应是指利用测序技术对dna或rna样本中的核苷酸序列进行确定。这个过程包括核酸模板的制备、核酸链的合成和数据的读取,最终输出生物样本的精确遗传信息。
64、利用扩增子片段构建文库:利用pcr技术特异性扩增目标dna区域(扩增子),然后将这些扩增子处理并整合到测序文库中。这种方法常用于目标基因的选择性测序。
65、纳米孔测序技术:一种单分子测序技术,通过检测dna分子通过蛋白质纳米孔时产生的电流变化来确定碱基序列。具有读长长、实时测序等特点。
66、方法优化:通过调整实验条件、改进实验步骤或分析算法等手段,提高方法的灵敏度、特异性、准确性或效率。
67、illumina平台的检测结果:使用illumina公司的测序仪获得的数据。通常指高通量、短读长的测序数据,具有高准确度的特点。
68、多个关键参数上进行了优化:对影响方法性能的多个重要因素进行调整和改进,包括pcr引物设计、反应条件、文库制备方法、测序深度、数据分析流程等。本发明的检测方法相较于现有技术具有显著的优势:
69、本发明提供了一种创新的结核分枝杆菌耐药性预测方法,通过长片段多重pcr扩增子测序策略,直接从临床样本中扩增与耐药相关的基因。与传统基于培养的耐药表型测试(pdst)和现有的分子诊断技术相比,此方法避免了操作复杂和耗时的问题,且能够检测更广泛的耐药突变,提供完整的药物敏感性信息。此外,市场上的其他下一代测序产品如deeplex myc-tb,虽然能够进行高覆盖率的靶向测序,但通常依赖于短扩增子,这可能限制了其在检测复杂基因区域或较大片段插入/缺失的能力,本发明通过采用长扩增子,不仅能够覆盖更广泛的基因组区域,还能提高对稀有变异的检测能力,增强了测试的全面性和精确度。本发明在数据处理上也展示了显著的优势,特别是在生物信息学分析流程的自动化和优化方面。通过使用先进的生物信息学工具和算法,本发明能够快速准确地从大量复杂的测序数据中提取有用信息,进行高效的数据解读和分析。本发明采用的数据处理流程包括自动化的错误校正和数据过滤步骤,这有助于提高最终结果的准确性和可靠性。通过对原始测序数据进行质量控制,自动去除低质量或偏离标准的数据,确保分析结果基于高质量的数据集。本发明的数据分析平台能够自动执行复杂的比对、突变检测和耐药性预测算法。这种高度自动化的流程减少了人工干预的需求,使得非专业人员也能轻松操作,从而降低了技术门槛和操作错误的可能。
70、本发明的数据分析流程专门为纳米孔靶向测序技术定制,形成了一个完整的一体化分析框架,目的是提高数据处理的效率和准确性。整个流程涵盖质量控制、变异识别、功能注释以及最终报告生成等关键步骤。在数据处理过程中,我们优化了包括读长覆盖、质量分数阈值和变异频率阈值在内的多个关键参数,确保即使在复杂基因区域和低测序深度的条件下,也能有效识别和确认关键变异。
71、流程首先进行严格的质量控制,排除低质量读取片段和非特异性对齐,保障数据的纯净度。然后,将高质量的读取片段准确地映射到参考基因组,识别潜在的变异点,并对识别出的变异进行详尽的生物学注释,分析其对蛋白质功能的可能影响,为临床决策提供科学依据。最终,系统自动整合所有分析结果,生成详细的耐药性预测报告,为医疗提供者提供可靠的决策支持。此一体化流程通过不断的技术优化和参数调整,确保了操作的高效率和数据的高准确性,从而显著提升了临床诊断和治疗的精度。
72、本发明所述的检测方法,重点在于以下步骤:
73、1)设计多重pcr反应引物和确定反应条件:
74、本发明针对与16种常用抗结核药物耐药性相关的关键基因(详见表1)进行了特定的引物设计。我们开发了一组多重pcr引物(详见表2),旨在全面覆盖已知的抗结核药物耐药位点。在设计这些多重pcr引物时,我们特别注重引物的特异性和全面性,以确保能够准确地扩增所有目标基因区域。此外,我们还精确设定了pcr的反应条件,以优化扩增效率和结果的可靠性,确保每次实验都能获得高质量的数据。
75、2)文库构建、测序反应的实验流程:
76、在使用纳米孔测序技术进行文库构建和测序反应时,我们面对的是一种相对较新的测序技术,它在实践中的经验尚不如成熟的二代测序技术广泛。特别是在利用扩增子片段构建文库方面,目前尚缺乏官方的推荐标准或成熟的操作流程。因此,我们需要依据预实验的结果来定制适合本研究的建库和测序策略。在选择合适的策略时,我们综合考虑了数据的准确性、实验的复杂度以及所需的时间效率等关键因素。鉴于纳米孔测序技术的特点和在结核病研究中的应用潜力,我们特别注重评估这些技术参数,确保所采用的实验方案既能满足研究需求,又能产生可靠和精确的结果。这一过程的谨慎设计和优化,对于提高耐药结核病检测的效率和质量至关重要。
77、3)生物信息学分析流程的建立:
78、在进行生物信息学分析时,虽然不同测序平台的基本处理流程相似,针对各平台生成的数据可能需要使用特定的软件或调整现有的流程。分析的首要步骤是将原始测序数据进行格式转换并按照条形码进行分类,这一步骤是确保后续分析准确性的关键。随后,进行数据的质量控制,剔除低质量的读取,以增强分析的可靠性。经过质控的数据将被用于比对分析,此时数据将对照参考基因组或特定的目标基因库进行比对。通过比对,可以将测序读取正确地分类到对应的基因。对于属于同一基因扩增子的读取进行互相校正,确保最终获得的序列数据的准确性和完整性。为实现这一复杂流程的高效执行,我们开发了自动化脚本,从而提升数据处理的速度和准确性。这些生物信息学分析步骤对于深入理解结核病相关基因的序列数据至关重要,能够为疾病机理的研究和治疗策略的制定提供精确的遗传信息。在分析过程中,我们还需考虑实验设计、样本处理和结果解释等多个方面,以确保研究的整体可靠性和科学性。
79、4)方法优化:
80、在我们的检测流程中,方法优化主要关注两个关键方面。首先,鉴于采用了多重pcr技术来同时扩增多个基因,存在扩增效率不均的问题。为了确保每个基因都能获得充分的扩增,我们将样品分成两组进行扩增。尽管如此,为了解决不同基因之间扩增片段丰度的显著差异,仍需进一步的优化。特别是通过调整引物的浓度,我们可以确保各个基因的扩增效率更加均匀,从而提高数据的均衡性和整体质量。
81、其次,为确保检测结果的准确性,我们将illumina平台的检测结果作为金标准,基于此对纳米孔测序平台的数据分析流程进行了参数优化,特别是在读长覆盖、质量分数阈值和变异频率阈值等多个关键参数上进行了优化,以提高纳米孔测序数据的处理质量。这些调整显著提升了整体测序方案的效率和精确度,同时也有效地降低了成本。通过这些优化步骤,我们为结核病耐药性研究提供了更加精确且可靠的遗传信息,从而支持该领域的科学研究和临床应用。
82、本发明的检测方法相较于现有技术具有显著的优势:
83、本发明提供了一种创新的结核分枝杆菌耐药性预测方法,通过长片段多重pcr扩增子测序策略,直接从临床样本中扩增与耐药相关的基因。与传统基于培养的耐药表型测试(pdst)和现有的分子诊断技术相比,此方法避免了操作复杂和耗时的问题,且能够检测更广泛的耐药突变,提供完整的药物敏感性信息。此外,市场上的其他下一代测序产品如deeplex myc-tb,虽然能够进行高覆盖率的靶向测序,但通常依赖于短扩增子,这可能限制了其在检测复杂基因区域或较大片段插入/缺失的能力,本发明通过采用长扩增子,不仅能够覆盖更广泛的基因组区域,还能提高对稀有变异的检测能力,增强了测试的全面性和精确度。本发明在数据处理上也展示了显著的优势,特别是在生物信息学分析流程的自动化和优化方面。通过使用先进的生物信息学工具和算法,本发明能够快速准确地从大量复杂的测序数据中提取有用信息,进行高效的数据解读和分析。本发明采用的数据处理流程包括自动化的错误校正和数据过滤步骤,这有助于提高最终结果的准确性和可靠性。通过对原始测序数据进行质量控制,自动去除低质量或偏离标准的数据,确保分析结果基于高质量的数据集。本发明的数据分析平台能够自动执行复杂的比对、突变检测和耐药性预测算法。这种高度自动化的流程减少了人工干预的需求,使得非专业人员也能轻松操作,从而降低了技术门槛和操作错误的可能。
- 还没有人留言评论。精彩留言会获得点赞!