一种新型含BODIPY类共轭单元有机染料敏化剂及其制备方法与流程
本发明涉及染料敏华太阳能电池材料领域,具体的涉及一种新型含BODIPY类共轭单元有机染料敏化剂。
背景技术:
:
自1991年瑞士小组首次采用联吡啶钌作为染料与纳米多孔TiO2薄膜制备了染料敏化太阳能电池(DSSCs)以来,染料敏化纳米晶TiO2太阳能电池就凭借低制备成本和较高的光电转换效率成为近二十年来太阳能光电转换领域的研究热点。BODIPY母核具有多个活性位点,可以根据热学、光学和电学性能要求进行适当的修饰和官能化。人们通过对BODIPY母核各个位点进行修饰,合成了许多性能优异的新型BODIPY染料。研究者们通常选择在BODIPY母体的两个吡咯环上进行修饰,通过引入不同性能的官能团,可以有效延长体系共轭链长,增加核心部分的刚性平面结构,其中在母核的3,5-位及2,6-位上引入取代基对染料分子的光谱性能影响最大。2,6-位修饰给受体电子基团的BODIPY染料敏化剂在2012年为Dai等报道,他们合成了以BODIPY为共轭π桥,苯并咪唑衍生物为给体单元,氰乙酸为吸附基团的一系列染料。该类分子表现了较宽的吸收光谱,能量转换效率最高达到了2.26%,这是当时基于BODIPY染料敏化电池器件的最高光电转换效率,展现了巨大的应用潜力。然而,关于这类2,6-位修饰的BODIPY染料敏化剂的效率一直未得到明显的提高。
三苯胺是另一类光电性能优异的电子传输材料,由于其具有强的给电子能力及原料价格低廉易得,已经越来越广泛应用于DSSC领域。然而其非平面的立体结构减弱了对光的吸收,因此我们将BODIPY单元与不同刚性的三苯胺衍生物结合起来,希望改善染料对光的吸收及光捕获效率,从而提高电池的性能。通过改变给体的刚性平面结构,研究不同给体单元的给电子能力大小对染料的光谱和能级的调控作用,从而改善电池性能参数。
技术实现要素:
:
本发明的目的是提供一种新型含BODIPY类共轭单元有机染料敏化剂,具有新颖的分子结构、有较低的能隙值、宽的光谱吸收范围,且其制备方法简单,合成率高。
本发明的另一个目的是提供该含BODIPY类共轭单元有机染料敏化剂的制备方法,其制备方法简单,产品纯化简单,产率高,且具有普适性。
为实现上述目的,本发明采用以下技术方案:
一种新型含BODIPY共轭单元的有机染料敏化剂,该物质具有通式(I)的化学结构:
式(I)中,D是给体单元,为以下结构单元中的一种:
其中,R、R1为H或CnH2n+1烷基或烷氧基,n为1-20的自然数。
一种新型含BODIPY共轭单元的有机染料敏化剂的制备方法,包括以下步骤:
(1)在碱的作用下,对羟基苯甲醛经烷基化反应,制得中间体1,其结构为:
(2)中间体1与吡咯缩合得到二吡咯烷中间体,二吡咯烷在三乙胺作用下与三氟化硼乙醚络合反应得到中间体2,其结构为:
(3)中间体2与一氯化碘经亲电取代反应,制得中间体3,其结构为:
(4)中间体3在四(三苯基膦基)钯的作用下与4-甲酰基苯硼酸经Suzuki偶联反应,制得中间体4,其结构为:
(5)中间体4再与一氯化碘经亲电取代反应,制得中间体5,其结构为:
(6)对烷氧基碘苯与对溴苯胺经过乌尔曼反应,得到中间体6,其结构为:
(7)中间体6在四(三苯基膦基)钯的催化作用下经Suzuki偶联反应,制得中间体7,其结构为:
(8)10-烷氧基亚氨基芪与对溴碘苯经Buchwald–Hartwig偶联反应,得到中间体8,其结构为:
(9)中间体8在四(三苯基膦基)钯的催化作用下经Suzuki偶联反应,得到中间体9,其结构为:
(10)将对烷氧基碘苯与2-溴咔唑在碘化亚铜催化下经过乌尔曼缩合反应,制得中间体10,其结构为:
(11)将中间体10与双联频哪醇硼酯经过Suziki偶联反应,制得中间体11,其结构为:
(12)中间体7、中间体9和中间体11分别与中间体5经Suzuki偶联反应,制得中间体12、13、14,其结构为:
D为
(13)中间体12、13、14分别在氰乙酸的作用下经Knoevenagel缩合反应,分别制得目标染料Dye1、2、3,其结构为:
作为上述技术方案的优选,步骤(1)-(13)中,所述反应的反应介质为乙腈、N,N-二甲基甲酰胺、甲醇、四氢呋喃、二氯甲烷、1,4-二氧六环、甲苯、氯仿、乙醇中的一种或几种混合。
作为上述技术方案的优选,步骤(2)、(6)、(8)、(11)、(12)和(13)中所述反应中,所用的催化剂为三氯化铟、四(三苯基膦基)钯、三(二亚苄基丙酮)二钯、氯化亚铜、碘化亚铜和哌啶中的一种。
作为上述技术方案的优选,步骤(1)、(4)、(6)、(7)、(8)、(9)、(10)、(11)、(12)和(13)所述反应中,所用的碱为碳酸钾、三乙胺、氢氧化钾、乙酸钾、哌啶和叔丁醇钠中的一种。
作为上述技术方案的优选,步骤(1)-(13)中,所述反应中的反应温度为25-120℃,反应时间为2-48h。
本发明具有以下有益效果:
(1)在合成方法上,该方法原料普通易得,生产成本低,反应条件更易控制。
(2)通过对染料的光谱数据分析,我们可以看出该类染料具有良好的光子捕获能力,相较于文献报道的染料具有宽的紫外吸收范围,应用于染料敏化太阳能电池,具有高的光电转换效率。
附图说明:
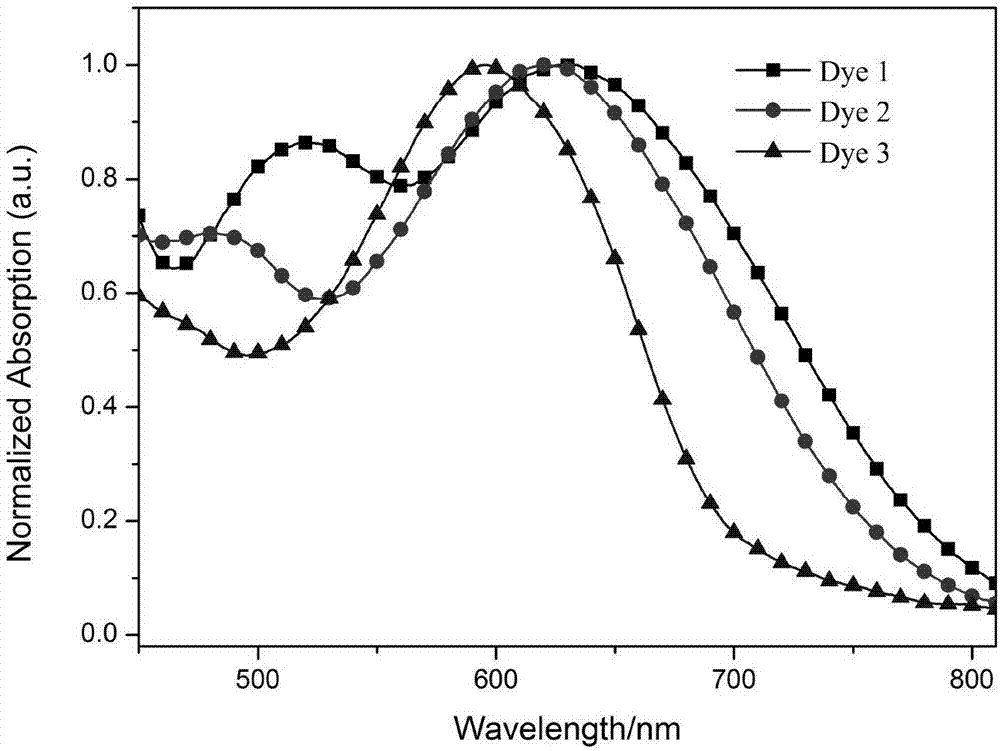
图1为Dye 1-3在二氯甲烷溶液中的紫外吸收光谱;
图2为Dye1-3在TiO2膜上归一化的紫外-可见吸收光谱;
图3为Dye 1的核磁氢谱;
图4为Dye 1的核磁碳谱;
图5为Dye 2的核磁氢谱;
图6为Dye 2的核磁碳谱;
图7为Dye 3的核磁氢谱;
图8为Dye 3的核磁碳谱;
图9为Dye 1-3在二氯甲烷溶液中的循环伏安曲线;
图10为Dye 1-3制备有机染料敏化太阳能电池的I-V曲线。
具体实施方式:
为了更好的理解本发明,下面通过实施例对本发明进一步说明,实施例只用于解释本发明,不会对本发明构成任何的限定。
(1)中间体1的合成
在500mL的圆底烧瓶中依次加入对羟基苯甲醛(12.5g,0.1mol)、碳酸钾(16.5g,0.12mol)、溴代正辛烷(23.2g,0.12mol)和150mL乙腈,磁力搅拌,将反应温度控制在80℃反应10h。停止反应,用抽滤漏斗过滤除掉固体残渣,旋干溶剂,用二氯甲烷稀释,大量水洗,无水硫酸镁干燥。过滤收集滤液,减压旋除溶剂,粗产品用硅胶(200-300目)柱层析[洗脱液,V(石油醚):V(乙酸乙酯)=10:1],纯化得到淡黄色液体中间体1(22.2g),产率95%。1H NMR(400MHz,CDCl3)δ:9.82(d,J=1.4Hz,1H),7.77(dd,J=8.7,1.5Hz,2H),6.93(dd,J=8.7,1.5Hz,2H),3.97(dd,J=6.6,1.6Hz,2H),1.82–1.65(m,2H),1.50–1.35(m,2H),1.35–1.18(m,10H),0.87(t,J=6.9Hz,3H).13C NMR(100MHz,CDCl3)δ:190.45,164.49,132.19,130.05,114.70,68.25,30.91,29.70,29.31,28.78,25.61,22.42,13.93.
(2)中间体2的合成
在100mL单口瓶中加入中间体1(2.34g,10mmol)和新蒸的吡咯(30mL,430mmol),向反应瓶中通氩气置换空气10min,在氩气保护下迅速加入催化剂InCl3(0.11g,0.5mmol),在室温下磁力搅拌5h,再向反应瓶中加入NaOH(0.2g,5mmol)粉末继续搅拌30min,淬灭反应。减压蒸馏,回收多余的吡咯,得二吡咯甲烷中间体,取二吡咯甲烷中间体(3.50g,10mmol)溶解于80mL二氯甲烷中,加入四氯苯醌(2.9g,12mmol),室温条件下磁力搅拌,充分氧化8h。然后将反应混合液置于氩气保护下,慢慢滴加37mL三氟化硼乙醚(300mmol),10min后再缓慢加入41.7mL三乙胺(300mmol),滴加完毕后继续反应8h后。将反应液倒入装有200-300目硅胶的柱子中粗过滤,以二氯甲烷为淋洗剂,有机相用饱和氢氧化钠溶液洗3次(100mL×3),收集有机相,无水硫酸钠干燥,过滤收集滤液,减压旋除溶剂,粗产品经硅胶(200-300目)柱层析[洗脱液,V(石油醚):V(乙酸乙酯)=10:1]纯化得墨绿色粉末状中间体2(2.50g),产率63%。1H NMR(400MHz,CDCl3)δ:7.92(s,2H),7.54(d,J=8.5Hz,2H),7.03(d,J=8.6Hz,2H),6.98(d,J=3.2Hz,2H),6.58–6.53(m,2H),4.06(t,J=6.4Hz,2H),1.86–1.82(m,2H),1.42–1.27(m,10H),0.89(t,J=7.0Hz,3H).13C NMR(100MHz,CDCl3)δ:162.11,147.42,142.95,134.79,132.76,131.51,126.00,118.26,114.84,68.25,30.91,29.70,29.31,28.78,25.61,22.42,13.93.
(3)中间体3的合成
向100mL三口瓶中依次加入中间体2(1.0g,2.5mmol)、40mL无水甲醇和40mL二氯甲烷,室温下磁力搅拌几分钟,抽真空,通入氩气保护,之后将ICl(0.49g,3.0mmol)溶解在5mL无水甲醇中,用注射器逐滴加入到反应瓶中,滴加完毕后再继续反应1h,整个反应过程避光。经TLC检测原料基本反应完,停止反应,向三口瓶中加入50mL蒸馏水,用二氯甲烷萃取三次(80mL×3),有机层再经饱和食盐水洗涤三次(100mL×3),合并有机相,无水硫酸镁干燥过夜。过滤收集滤液,减压蒸除溶剂,得到的粗产品用硅胶(200-300目)柱层析[洗脱液,V(石油醚):V(二氯甲烷)=20:1]纯化得红色固体中间体3(0.78g),产率60%。1H NMR(400MHz,CDCl3)δ:7.97(s,1H),7.82(s,1H),7.52(d,J=8.5Hz,2H),7.04(d,J=8.7Hz,4H),6.61(s,1H),4.06(t,J=6.4Hz,2H),1.87–1.82(m,2H),1.42–1.31(m,10H),0.89(t,J=7.0Hz,3H).13C NMR(100MHz,CDCl3)δ:162.16,146.83,146.00,145.04,135.93,135.77,135.04,132.91,132.53,125.68,119.30,114.78,68.47,31.84,29.34,29.25,29.15,26.05,22.68,14.12.
(4)中间体4的合成
向100mL三口瓶中依次加入中间体3(0.26g,0.5mmol)、4-甲酰基苯硼酸(0.12g,0.75mmol)、20mL四氢呋喃装上回流冷凝管,抽真空,置换氩气保护;之后加入催化量的四(三苯基膦)钯和K2CO3(2M,3mL)溶液,升温至80℃回流过夜;停止反应,冷却至室温,将反应液倒入100mL蒸馏水中,用二氯甲烷萃取3次(60mL×3),合并有机相,有机相饱和食盐水洗三次(100mL×3),无水硫酸镁干燥过夜。过滤收集滤液,减压旋除溶剂,得到的粗产物经硅胶(300-400目)柱层析[洗脱液,V(石油醚):V(二氯甲烷)=5:1]纯化得红褐色粉末中间体4(0.19g),产率78%。1H NMR(400MHz,CDCl3)δ:9.99(s,1H),8.29(s,1H),8.00(s,1H),7.89(d,J=8.0Hz,2H),7.69(d,J=8.0Hz,2H),7.59(d,J=8.5Hz,2H),7.22(s,1H),7.10(s,1H),7.07(d,J=7.2Hz,2H),6.62(d,J=2.5Hz,1H),4.08(t,J=6.4Hz,2H),1.88–1.84(m,2H),1.40–1.28(m,10H),0.90(t,J=6.9Hz,3H).13C NMR(100MHz,CDCl3)δ:191.49,162.12,144.95,140.20,139.01,135.49,135.09,132.53,131.98,130.54,125.86,125.65,119.13,114.80,68.49,31.84,29.35,29.26,29.18,26.07,22.68,14.11.
(5)中间体5的合成
在50mL三口瓶中加入中间体4(0.4g,0.8mmol)、10mL无水甲醇和10mL DMF,在室温下磁力搅拌几分钟后,抽真空,置换氩气保护。之后将ICl(0.16g,1.0mmol)溶解在5mL无水甲醇中,用注射器逐滴加入到反应瓶中,5min滴加完毕,继续反应2h,整个过程避光。经TLC检测原料反应完,停止反应,向三口瓶中加入20mL饱和硫代硫酸钠溶液。继续搅拌15min,反应液用二氯甲烷萃取3次(30mL×3),有机相饱和食盐水洗涤2次(100mL×2),最后经无水硫酸镁干燥过夜。过滤收集滤液,减压旋除溶剂,粗产物经硅胶(300-400目)柱层析[洗脱液,V(石油醚):V(二氯甲烷)=8:1]纯化得到深红色粉末固体中间体5(0.44g),产率89%。1H NMR(400MHz,CDCl3)δ:10.00(s,1H),8.33(s,1H),8.00(d,J=7.9Hz,1H),7.90(d,J=7.4Hz,2H),7.69(d,J=7.8Hz,2H),7.58(d,J=8.2Hz,2H),7.27(s,1H),7.15(s,1H),7.09(d,J=8.3Hz,2H),4.09(t,J=6.3Hz,2H),1.90–1.84(m,2H),1.42–1.29(m,10H),0.91(s,3H).13C NMR(100MHz,CDCl3)δ:191.42,162.48,147.46,141.89,138.44,136.94,135.34,132.60,130.55,130.37,128.04,127.15,125.75,125.49,115.01,68.56,31.84,29.34,29.26,29.15,26.06,22.68,14.12.
(6)中间体6的合成
向100mL三口瓶中依次加入对碘苯甲醚(1.1g,4.8mmol)、对溴苯胺(0.34g,2.0mmol)、氢氧化钾(0.56g,10mmol)、1,10-菲啰啉(0.03g,0.16mmol)和60mL甲苯,装上回流冷凝管,抽真空,置换氩气保护。在氩气流下,迅速加入催化量的氯化亚铜,升温至回流反应20h。经TLC检测原料反应完,停止反应,减压蒸除溶剂,将反应混合物溶于100mL二氯甲烷,再经饱和食盐水洗涤3次(80mL×3),有机相经无水硫酸钠干燥过夜。过滤收集滤液,减压旋除溶剂,粗产物经硅胶(200-300目)柱层析[洗脱液,V(石油醚):V(乙酸乙酯)=8:1]纯化得到淡黄色固体中间体6(0.60g),产率78%。1H NMR(400MHz,CDCl3)δ:7.31(d,J=9.0Hz,2H),7.04–7.02(m,4H),6.93–6.91(m,4H),6.67(d,J=9.0Hz,2H),3.74(s,6H).
(7)中间体7的合成
向100mL三口瓶中依次加入中间体6(0.76g,2.0mmol)、双联频哪醇硼酯(0.76g,3.0mmol)、乙酸钾(0.78g,8.0mmol)和40mL1,4-二氧六环。抽真空,置换氩气保护,加入催化量的Pd(dppf)Cl2。将温度控制在85℃反应24h,停止反应,冷却至室温。将反应液倒入100mL蒸馏水中,二氯甲烷萃取3次(80mL×3),合并有机相,有机相再经饱和食盐水洗涤3次(100mL×3),无水硫酸镁干燥过夜。过滤收集滤液,减压蒸除溶剂,粗产物经硅胶(200-300目)柱层析[洗脱液,V(石油醚):V(乙酸乙酯)=10:1]纯化得到黄色粉末固体中间体7(0.65g),产率75%。1H NMR(400MHz,CDCl3)δ:7.60(d,J=7.8Hz,2H),7.06(d,J=8.2Hz,4H),6.86(d,J=8.3Hz,2H),6.83(d,J=8.3Hz,4H),3.80(s,6H),1.32(s,12H).13C NMR(100MHz,CDCl3)δ:156.12,140.33,135.73,127.12,118.55,114.66,83.39,55.45,24.82.
(8)中间体8的合成
在100mL三口瓶中依次加入10-甲氧基亚氨基芪(0.67g,3.0mmol)、对溴碘苯(1.3g,4.5mmol)、叔丁醇钠(0.58g,6mmol)和30mL甲苯。装上回流冷凝管,抽真空,置换氩气保护,之后迅速加入催化量的Pd2(dba)3和3-叔丁基膦,升温至回流反应8h。停止反应,冷却至室温,将反应液倒入100mL蒸馏水中,二氯甲烷萃取3次(60mL×3),有机相再经饱和食盐水洗涤3次(100mL×3),无水硫酸钠干燥过夜。过滤收集滤液,减压旋除溶剂,粗产物经硅胶(200-300目)柱层析[洗脱液,V(石油醚):V(乙酸乙酯)=20:1]纯化得到淡黄色油状液体中间体8(0.62g),产率54%。1H NMR(400MHz,CDCl3)δ:7.81(dd,J=7.9,1.4Hz,1H),7.55–7.50(m,1H),7.45–7.32(m,5H),7.30(dd,J=7.4,1.5Hz,1H),7.10–7.04(m,2H),6.27–6.21(m,2H),6.03(s,1H),3.78(s,3H).
(9)中间体9的合成
向100mL三口瓶中依次加入中间体8(1.13g,3.0mmol)、双联频哪醇硼酯(1.14g,4.5mmol)、乙酸钾(1.18g,12.0mmol)和40mL1,4-二氧六环。用类似合成中间体7的方法得到浅黄色固体中间体9(0.87g),产率68%。1H NMR(400MHz,CDCl3)δ:7.79(d,J=7.7Hz,1H),7.52–7.42(m,5H),7.37(d,J=7.2Hz,3H),7.29(d,J=7.3Hz,1H),6.39(d,J=8.4Hz,2H),6.02(s,1H),3.76(s,3H),1.27(s,12H).13C NMR(100MHz,CDCl3)δ:156.12,150.60,142.63,135.87,135.70,134.22,130.74,130.01,129.46,129.43,128.32,127.52,127.17,126.98,110.98,102.15,83.17,55.32,24.77.
(10)中间体10的合成
向100mL三口瓶中依次加入2-溴咔唑(2.45g,10mmol),对碘苯甲醚(2.58g,11mmol),碘化亚铜(190mg,1mmol),L-脯氨酸(230mg,2mmol),碳酸钾(2.76g,20mmol)和30mL二甲基亚砜。升温至90℃反应40h。停止反应,冷却至室温,加100mL蒸馏水稀释,混合液用乙酸乙酯萃取3次(60mL×3),有机相经饱和食盐水洗涤3次(100mL×3),无水硫酸镁干燥。过滤收集滤液,粗产品经硅胶柱层析,得到灰白色固体中间体10(2.3g),产率65%。1H NMR(400MHz,CDCl3)δ:8.13(d,J=7.1Hz,1H),8.01(d,J=7.9Hz,1H),7.51–7.38(m,5H),7.32(d,J=7.9Hz,2H),7.15(d,J=7.1Hz,2H),3.95(s,3H).
(11)中间体11的合成
向100mL三口瓶中,依次加入中间体10(3.51g,10mmol)、双联频哪醇硼酯(3.81g,15mmol)。用类似合成中间体7的方法得到白色晶体中间体11(2.7g),产率67%。1H NMR(400MHz,CDCl3)δ:8.19–8.13(m,2H),7.80–7.72(m,2H),7.46(d,J=8.7Hz,2H),7.41(d,J=7.4Hz,1H),7.30(d,J=8.6Hz,2H),7.13(d,J=8.6Hz,2H),3.94(s,3H),1.36(s,12H).
(12)中间体12的合成
向100mL三口瓶中依次加入中间体5(0.31g,0.5mmol)、中间体7(0.26g,0.6mmol)、30mL四氢呋喃和碳酸钾溶液(2M,3mL),装上回流冷凝管,抽真空,置换氩气保护。再向反应瓶中加入催化量的四(三苯基膦基)钯,升温至80℃反应24h。停止反应,冷却至室温,将反应液倒入100mL蒸馏水中,二氯甲烷萃取3次(60mL×3),有机相再用饱和食盐水洗涤3次(100mL×3),无水硫酸镁干燥过夜。过滤收集滤液,减压旋除溶剂,粗产物经硅胶(200-300目)柱层析[洗脱液,V(石油醚):V(乙酸乙酯)=5:1]纯化得到墨绿色固体中间体12(0.35g),产率80%。1H NMR(400MHz,CDCl3)δ:9.98(s,1H),8.30(s,1H),8.24(s,1H),7.88(d,J=8.1Hz,2H),7.68(d,J=8.1Hz,2H),7.61(d,J=8.5Hz,2H),7.34(d,J=8.6Hz,2H),7.17(s,1H),7.11(s,1H),7.08(s,6H),6.92(d,J=8.6Hz,2H),6.84(d,J=8.8Hz,4H),4.09(t,J=6.4Hz,2H),3.80(s,6H),1.89–1.84(m,2H),1.38–1.32(m,10H),0.90(t,J=5.9Hz,3H).13C NMR(100MHz,CDCl3)δ:191.56,161.95,156.11,148.62,146.22,143.74,140.49,139.24,139.15,136.45,135.44,135.23,134.89,132.44,130.53,126.75,126.15,125.97,125.54,124.96,124.58,123.98,120.39,114.78,110.03,103.82,68.45,55.49,31.83,29.34,29.25,29.18,26.06,22.67,14.11.MALDI-TOF-MS,m/z:calcd for C50H48BF2N3O4:803.370,found:803.474[M]+.
(13)中间体13的合成
向100mL三口瓶中依次加入中间体5(0.31g,0.5mmol)、中间体9(0.25g,0.6mmol)。用类似合成中间体12的方法得到绿色固体中间体13(0.30g),产率74%。1H NMR(400MHz,CDCl3)δ:9.98(s,1H),8.23(s,2H),7.87–7.82(m,3H),7.68(s,2H),7.59–7.54(m,3H),7.47(d,J=5.7Hz,2H),7.42–7.36(m,3H),7.31(d,J=8.1Hz,1H),7.21(s,2H),7.14(s,1H),7.08(s,2H),6.99(s,1H),6.42(s,2H),6.05(s,1H),4.08(d,J=2.3Hz,2H),3.79(s,3H),1.89–1.86(m,2H),1.35–1.29(m,10H),0.90(s,3H).13C NMR(100MHz,CDCl3)δ:191.54,161.86,156.19,148.33,145.85,144.04,142.82,141.12,138.71,136.52,135.89,135.33,134.84,134.27,132.42,130.81,130.51,130.12,129.51,128.43,127.62,127.28,127.11,126.04,125.50,124.65,124.15,121.86,114.74,112.22,106.50,102.27,68.42,55.38,31.83,29.35,29.25,29.17,26.06,22.68,14.12.MALDI-TOF-MS,m/z:calcd.for C51H46BF2N3O3:797.360,found:797.169[M]+.
(14)中间体14的合成
向100mL三口瓶中依次加入中间体5(0.31g,0.5mmol)、中间体11(0.24g,0.6mmol)。用类似合成中间体12的方法得到绿色固体中间体14(0.30g),产率76%。1H NMR(400MHz,CDCl3)δ:9.99(s,1H),8.38(s,1H),8.28(s,1H),8.12(dd,J=7.8,2.9Hz,2H),7.89(d,J=8.3Hz,2H),7.69(d,J=8.2Hz,2H),7.64(d,J=8.7Hz,2H),7.48–7.43(m,4H),7.42–7.38(m,1H),7.30(dd,J=8.0,3.6Hz,2H),7.23(s,1H),7.20(s,1H),7.17(d,J=2.1Hz,1H),7.15(d,J=2.1Hz,1H),7.13(s,1H),7.11(s,1H),4.10(t,J=6.5Hz,2H),3.96(s,3H),1.89–1.86(m,2H),1.54–1.36(m,10H),0.91(t,J=6.9Hz,3H).13C NMR(100MHz,CDCl3)δ:191.60,162.08,159.13,146.89,143.78,142.11,141.91,139.74,139.08,136.27,134.98,132.53,131.70,130.58,130.09,129.85,128.71,126.18,125.89,125.59,122.88,122.79,120.90,120.28,120.02,117.72,115.37,114.87,109.85,106.33,77.38,77.06,76.74,68.49,55.67,31.87,29.40,29.30,29.21,26.11,22.73,14.18.MALDI-TOF-MS,m/z:calcd.for C49H44BF2N3O3:771.340,found:771.214[M]+.
实施例1
Dye 1的合成:
在100mL三口瓶中依次加入中间体12(0.16g,0.2mmol)、氰乙酸(0.04g,0.4mmol)、20mL氯仿和20mL乙腈。装上回流冷凝管,抽真空,置换氩气保护,迅速加入2滴哌啶,升温至80℃回流反应24h。停止反应,冷却至室温,加入100mL二氯甲烷稀释反应液,有机相饱和食盐水洗涤3次(100mL×3),无水硫酸镁干燥过夜。过滤收集滤液,减压旋除溶剂,粗产物经硅胶(300-400目)柱层析[洗脱液,V(二氯甲烷):V(甲醇)=10:1]纯化得到深绿色固体粉末Dye1(0.14g),产率80%。1H NMR(400MHz,DMSO-d6)δ:8.74(d,J=4.4Hz,2H),7.94–7.88(m,5H),7.73(d,J=8.5Hz,2H),7.63(d,J=8.6Hz,2H),7.43(s,1H),7.26(s,1H),7.19(d,J=8.5Hz,2H),7.03(d,J=8.8Hz,4H),6.92(d,J=8.9Hz,4H),6.74(d,J=8.7Hz,2H),4.11(t,J=6.3Hz,2H),3.74(s,6H),1.80–1.75(m,2H),1.45–1.28(m,10H),0.86(t,J=6.2Hz,3H).13C NMR(100MHz,DMSO-d6)δ:163.53,161.92,156.37,154.18,152.63,148.46,146.01,141.13,140.28,135.37,133.38,130.56,130.12,127.28,126.97,126.95,126.15,125.81,125.43,120.52,119.65,119.57,117.14,115.47,115.41,109.27,103.34,68.37,55.73,31.73,29.49,29.15,29.08,26.02,22.56,14.44.MALDI-TOF-MS,m/z:calcd.for C53H49BF2N4O5:870.380,found:870.196[M]+.
实施例2
Dye 2的合成:
在100mL三口瓶中依次加入中间体13(0.16g,0.2mmol)、氰乙酸(0.04g,0.4mmol)。用类似合成Dye1的方法得到绿色固体粉末Dye2(0.13g),产率78%。1H NMR(400MHz,DMSO-d6)δ:8.69(d,J=17.6Hz,1H),7.94–7.86(m,3H),7.78(d,J=8.2Hz,1H),7.74–7.69(m,1H),7.65(dd,J=10.0,4.4Hz,2H),7.56–7.38(m,7H),7.35–7.27(m,4H),7.18(dd,J=11.7,3.0Hz,3H),7.09–7.02(m,2H),6.27(d,J=4.0Hz,1H),6.22(d,J=9.0Hz,1H),4.12(t,J=8.1Hz,2H),3.77(d,J=7.3Hz,3H),1.81–1.76(m,2H),1.46–1.28(m,10H),0.88(s,3H).13C NMR(100MHz,DMSO-d6)δ:163.74,161.36,155.86,149.96,147.29,143.78,142.28,140.98,140.15,137.41,135.99,135.06,134.22,133.33,131.61,130.71,130.11,129.94,128.27,127.95,127.64,126.02,125.83,124.14,115.36,110.97,102.87,68.39,55.94,31.72,29.50,29.18,29.05,26.01,22.57,14.43.MALDI-TOF-MS,m/z:calcd.for C54H47BF2N4O4:864.370,found:864.212[M]+.
实施例3
Dye3的合成:
在100mL三口瓶中依次加入中间体14(0.16g,0.2mmol)、氰乙酸(0.04g,0.4mmol)。用类似合成Dye1的方法得到绿色固体粉末Dye3(0.13g),产率75%。1H NMR(400MHz,DMSO-d6)δ:8.82(s,1H),8.28–8.22(m,2H),8.02–7.93(m,4H),7.78(d,J=8.6Hz,1H),7.72–7.69(m,1H),7.56(d,J=8.8Hz,1H),7.49(s,1H),7.42(t,J=7.8Hz,4H),7.31(d,J=7.8Hz,4H),7.27–7.21(m,4H),7.06–7.03(m,1H),4.14(t,J=6.3Hz,2H),3.97–3.79(m,3H),1.85–1.71(m,2H),1.52–1.28(m,10H),0.91(t,J=6.9Hz,3H).13CNMR(100MHz,DMSO-d6)δ:163.47,162.03,159.03,146.70,143.85,141.84,141.62,139.42,139.14,135.38,133.47,132.46,132.06,130.76,130.12,129.93,128.87,126.25,125.86,124.06,122.80,122.51,121.45,120.92,120.43,118.67,115.83,115.45,110.04,106.82,68.38,55.90,31.74,29.48,29.17,29.10,26.02,22.58,14.45.MALDI-TOF-MS,m/z:calcd.for C52H45BF2N4O4:838.350,found:838.991[M]+.
上述实施例中目标染料Dye 1~3在二氯甲烷溶液中的紫外吸收光谱和电化学性质的相关数据分别见表1和表2;实施例中目标染料Dye 1~3的光电性能参数见表3。
表1 Dye 1~3的光物理性质
a Maximum absorption in DCM solution(10-5).
bεis the molar extinction coefficient atλmax of the maximum absorption.
c Maximum absorption on TiO2film.
由图1及表1数据可看出,三种染料在整个紫外-可见光区(300-850nm)都有覆盖,此外3种染料都表现出了3个明显的吸收峰。其中能量较高的吸收峰位于近紫外区300-400nm,这主要归因于电子在三苯胺衍生物芳环上产生的π-π*跃迁,属于给体单元的本征迁移吸收峰;染料在450-520nm处的吸收峰属于BODIPY共轭单元的特征吸收,归因于BODIPY单元的π-π*,S0-S2迁移;能量较低的一组吸收峰分别位于627nm、605nm和590nm,这主要是由于BODIPY单元的π-π*,S0-S1迁移和给体单元与受体单元之间的分子内电荷迁移(ICT)共同引起的。相较于染料Dye 2和Dye 3,染料Dye1表现出了22和37nm的红移现象,这说明了不同给体单元的给电子能力不同,即4,4-二甲氧基三苯胺表现出了强给电子能力,增强了给受体单元间电子转移,从而使λmax向长波方向移动。
由图2可看出,三种染料在固体TiO2膜上显示出了良好的集光性能,最大吸收峰分别位于632nm、621nm和595nm,相对于溶液中的吸收光谱,分别表现出了5nm、15nm和5nm的红移现象,这说明了染料在二氧化钛膜上发生了J-聚集。此外,相对于溶液中的吸收,三个染料分子在膜上的吸收光谱均表现出了一定程度的拓宽,表明了染料能够有效吸附于TiO2膜,这有利于提高染料对太阳光的捕获效率。
表2 Dye 1~3的电化学性质
[a]E0,0was estimated from the absorption thresholds from absorption spectra of dyes adsorbed on the TiO2film,E0,0=1240/λedgeabs.
[b]Eox was measured in CH2Cl2and calibrated by addition of 0.63V to the potential versus Fc/Fc+.
[c]Computed from the formula E*ox=EOX-E0,0.
从表2及图9中可看出,3种染料的基态氧化电位(Eox vs.NHE)均比I3-/I-电对的氧化/还原电位(0.42V vs.NHE)更正,即失去电子的氧化态染料分子能够有效被I-还原;染料分子的激发态还原电位均比二氧化钛的导带的能级(-0.5V vs.NHE)更负,才能使激发态的电子有效注入到半导体的导带,这样基于染料电池的电子循环得到了充分的利用,因而这些BODIPY染料可以作为二氧化钛电极的敏化剂。
表3 Dye 1~3的光电性能参数
从表3及图10可看出,三种染料的Jsc值分别为8.21,7.35和5.77mA·cm-2,相较于染料Dye2和Dye3,Dye1的敏化电池器件性能呈现出了最好的光伏性能,其短路电流密度Jsc为8.21mA·cm-2,开路电压为595mV,填充因子是0.64,对应的光电转换效率PCE达到3.12%。Dye1的短路电流密度最大主要归因于其具有更好的光捕获能力,对应了图1中其具有宽的光谱吸收和高的摩尔消光系数。染料Dye1~3中,Dye3的Voc值最高,这可能是因为染料Dye3的HOMO能级最低,染料再生的驱动力更足,降低了电子回传速率。
本发明通过上述实施例来说明详细合成方法,但本发明并不局限于上述方法,即不意味着本发明必须依赖上述反应条件才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明反应溶剂催化剂的等效替换及反应具体条件的改变等,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种染料敏化太阳能装置的制作方法
- 一种三维染料敏化太阳能电池工作电极及其制备方法与流程
- 一种BiIO敏化的BiIO/TiO2复合电极材料及其制备方法和应用与流程
- 一种可用染料敏化太阳能充电的复合型锂离子二次电池的制作方法与工艺
- 一种包含优化结构光电极的染料敏化太阳能电池的制造方法与工艺
- 敏化染料染色液及光电极的制造方法与流程
- 一种含BODIPY类共轭单元有机染料敏化剂及其制备方法与流程
- 低照度用染料敏化光电转换元件的电解质、以及使用该电解质的低照度用染料敏化光电转换元件的制造方法与工艺
- 一种具有染料敏化TiO2可见光催化降解气体甲醛的装置及方法与制造工艺
- 金属有机染料、染料敏化电极、太阳能电池和反应器的制作方法