生产光致发光颗粒的方法与流程
本申请涉及制造光致发光材料的颗粒的方法。
背景技术:
光致发光材料,也被称为磷光体,当其被第一波长的光激发时,能够发射不同于第一波长的第二波长的光。光致发光材料特别用于制备荧光涂层,尤其是制造显示屏、投影仪,特别是等离子屏、用于液晶显示器背光的灯、发光二极管、等离子灯、三色灯等。
光致发光材料的一个例子是由三价铈离子激活的钇铝石榴石(yag)(y3al5o12:ce3+),也称为yag:ce或yag:ce3+。这种光致发光材料特别用于在与蓝色发光二极管(led)联合后产生白光。为实现这一点,用包含yag:ce3+颗粒的涂层覆盖蓝色led。部分蓝光被光致发光涂层转换成黄光,这样能够获得白光。
光致发光材料可以通过固态反应制造。例如,在yag:ce的情况中,将铝的、钇的和铈的固体前体以粉末的形式混合,研磨,并在高温下加热(例如加热至高于1600℃的温度),以形成具有所需组成和晶相的颗粒的粉末。然后在还原气氛下,通常在氢气(h2)下,将粉末进行退火,以将无光致发光性质且充当电荷载体的受体的ce4+离子还原为具有所需光致发光性质的ce3+离子。所获得的光致发光颗粒具有良好质量的晶体结构。然后可以将它们分散在基质中(例如树脂中)以形成光致发光涂层。
包括固态反应的制造光致发光材料颗粒的方法能够制造平均尺寸大于1微米的,例如从10μm到15μm不等的颗粒。对于某些应用,期望制造平均尺寸小于1μm的颗粒,此类颗粒在下文中被无差别地称为纳米颗粒、纳米范围颗粒或亚微米颗粒。当需要形成包含光致发光颗粒的流体或粘性组合物以实施涂覆光致发光颗粒的所谓附加方法时尤其如此,例如通过在所需位置直接印刷包含光致发光颗粒的组合物。然而,形成包含随时间稳定的光致发光颗粒的流体组合物需要使用纳米颗粒。
为了制造纳米颗粒,已知实施所谓的自顶向下合成方法,其包括将通过固态反应获得的平均尺寸大于1微米的光致发光材料的颗粒进行研磨,例如在球磨机中研磨,以降低平均粒径。然而,研磨操作引起在所获得的颗粒的表面处形成缺陷,这导致光致发光纳米颗粒的光致发光性能的降低,特别是颗粒的外量子效率的降低。此外,可以观察到通过研磨获得的纳米范围颗粒的团聚现象,这引起大尺寸的颗粒簇的形成。
已知通过所谓的自下而上法形成光致发光材料的颗粒,其能够获得亚微米颗粒。自下而上合成法是化学合成的方法,其基于小尺寸的化学实体(原子或分子)的组装以形成较大尺寸的物体,在本文所述情形下为纳米颗粒。在自下而上法中,例如可以提及溶胶-凝胶法和溶剂热法或水热法。自下而上法的共同点是这一事实:纳米颗粒合成通常在低于固态反应温度的温度下进行。由于如此低的合成温度,光致发光纳米颗粒通常具有低结晶度,导致常常与电荷载体或发光“陷阱”相关的结构缺陷。因此,通过这种方法获得的光致发光纳米颗粒的光效率方面的性能远低于通过固态反应制备的平均尺寸大于1微米的光致发光颗粒的性能。
期望获得光致发光材料的纳米颗粒的粉末,其性能,特别是在光效率方面,高于通过自下而上法获得的相同光致发光材料的纳米颗粒所获得的性能。
技术实现要素:
一个实施方案的一个目的旨在克服前述的光致发光颗粒制造方法的全部或部分缺陷。
一个实施方案的另一个目的是使所述平均粒径小于1μm。
一个实施方案的另一个目的是使100%的所述颗粒(d100)具有小于1μm的尺寸。
一个实施方案的另一个目的包括补救在研磨步骤之后获得的纳米颗粒的表面缺陷。
一个实施方案的另一个目的是使纳米范围颗粒的外量子效率大于60%,并且内量子效率至少等于70%。
一个实施方案的另一个目的是允许形成在时间上稳定的胶体分散体。
一个实施方案的另一个目的是使合成的纳米颗粒的表面与用于形成磷光体的溶剂和/或包封剂相容。
一个实施方案的另一个目的是使光致发光纳米颗粒的分散体与在发光二极管上沉积磷光体的方法相容。
一个实施方案的另一个目的是使纳米颗粒在光敏树脂组合物中形成稳定的悬浮液。
一个实施方案的另一个目的是使纳米颗粒在丙烯酸酯型光敏树脂的组合物中形成稳定的悬浮液。
因此,一个实施方案提供了一种制造光致发光材料的纳米颗粒的方法,包括以下相继步骤:
a)形成所述光致发光材料的纳米范围颗粒;
b)在非水溶剂中形成含有所述颗粒的分散体,所述分散体还含有至少一种表面剂;
c)将所述分散体置于压力在2mpa至100mpa范围内的高压釜中;和
d)回收所述颗粒。
根据一个实施方案,在步骤b)之前,通过二氧化硅前体对所述纳米范围颗粒进行表面处理。
根据一个实施方案,其中所述表面剂是硅烷型偶联剂,该硅烷型偶联剂是具有如下化学式的有机官能化合物:
rnsix4-n
其中n等于1、2或3,x表示可水解的基团,且r是不可水解的有机基团。
根据一个实施方案,x是烷氧基、卤素基团或胺基。
根据一个实施方案,所述非水溶剂是醇。
根据一个实施方案,步骤a)包括:研磨平均尺寸大于1μm的所述光致发光材料的颗粒以获得所述纳米范围颗粒。
根据一个实施方案,其中所述光致发光材料的平均尺寸大于1μm的颗粒在湿环境中研磨。
根据一个实施方案,所述光致发光材料的平均尺寸大于1μm的颗粒在不同于步骤b)中使用的所述非水溶剂的溶剂中研磨。
根据一个实施方案,步骤c)的持续时间在30分钟至48小时的范围内。
根据一个实施方案,所述高压釜中的温度在25℃至300℃的范围内。
根据一个实施方案,所述光致发光材料是铝酸盐、硅酸盐、氮化物、氮氧化物、氟化物或硫化物。
根据一个实施方案,所述光致发光材料主要包括钇铝氧化物或镥铝氧化物,其还包含以下元素中的至少一种:铈、铕、铬、钕、铽、镝、镨或钆。
根据一个实施方案,所述方法还包括:在步骤c)之前,在用于形成所述纳米范围颗粒的全部或部分前体存在的情况下在高压釜中混合和处理所述纳米范围颗粒的步骤。
根据一个实施方案,所述方法还包括:在步骤c)之前,将所述纳米范围颗粒与至少一种光致发光物质混合的步骤。
根据一个实施方案,所述光致发光物质是量子点。
根据一个实施方案,所述纳米范围颗粒是量子点。
附图说明
前述和其他特征及优点将在以下特定实施方案的非限制性描述中结合附图进行详细讨论,其中:
图1以方框图的形式示出了制造光致发光材料的纳米范围颗粒的方法的一个实施方案;
图2是高压釜的局部简化截面图;
图3是通过图1中所示方法获得的光致发光材料的纳米颗粒的局部简化截面图;
图4是图3中所示的纳米颗粒的一部分的细节的局部简化截面图;
图5至7是通过图1中所示方法的变型获得的光致发光材料的颗粒的局部简化截面图;
图8以方框图的形式示出了图1中所示的纳米颗粒制造方法的一个步骤的一个更详细的实施方案;
图9示意性地示出了图1中所示方法的一个实施方案的各步骤中的纳米颗粒;
图10至12是光致发光颗粒的粒度分布曲线;
图13示出了光致发光强度根据由光致发光材料发射的辐射波长变化的曲线;
图14是代表光致发光纳米颗粒的一个实施例的x射线衍射图;
图15至18是光致发光颗粒粉末的粒度曲线;
图19示出了光致发光强度根据由光致发光材料发射的辐射波长变化的曲线;
图20、21和22是分别与图17、18和19相似的另一种光致发光材料的图;
图23、24和25是分别与图17、18和19相似的另一种光致发光材料的图;
图26、27和28是分别与图17、18和19相似的另一种光致发光材料的图;和
图29至32是光致发光颗粒粉末的粒度曲线。
具体实施方式
为清楚起见,在不同附图中相同的元件已经用相同的附图标记表示。此外,在以下描述中,表述“基本上”、“附近”和“约”表示“在10%以内”。
术语“材料的颗粒”表示该材料的单位元素。在本申请的上下文中所使用的术语“颗粒”应该在广义上来理解,并且其不仅对应于具有或多或少球形的致密颗粒,而且对应于角状颗粒、扁平颗粒、片状颗粒、纤维状颗粒或含纤维颗粒等。应理解,本发明上下文中的颗粒的“尺寸”是指颗粒的最小横向尺寸,例如,在纤维状颗粒的情况下,颗粒尺寸对应于纤维的直径。
根据本发明,术语“平均尺寸”是指在颗粒分布中大于50体积%的颗粒的尺寸且小于50体积%的颗粒的尺寸的颗粒尺寸。这对应于d50。因此,小于分布中100体积%的颗粒的尺寸的平均尺寸对应于d100。微米范围颗粒是指平均尺寸在1μm至100μm范围内,通常为1μm至50μm的颗粒。纳米颗粒是指平均尺寸小于1μm,优选5nm至500nm范围内的颗粒。微米范围颗粒的粒度可以通过例如使用malvernmastersizer2000进行激光衍射分析来测量。亚微米颗粒或纳米颗粒的粒度可以通过使用例如malvernzetasizernanozs进行动态光散射(dls)测量。
制造光致发光材料的纳米颗粒的方法的一个实施方案包括:用至少一种表面剂在非水溶剂中形成纳米颗粒的分散体,并将所述分散体在温度为50℃至300℃范围内且压力为2mpa至100mpa、优选2mpa至10mpa范围内的高压釜中保持30分钟至48小时。所述纳米范围颗粒可以通过对固态制造方法所获得的平均尺寸大于1μm的颗粒进行研磨来获得。作为变型,所述纳米颗粒可以通过自下而上法直接形成。
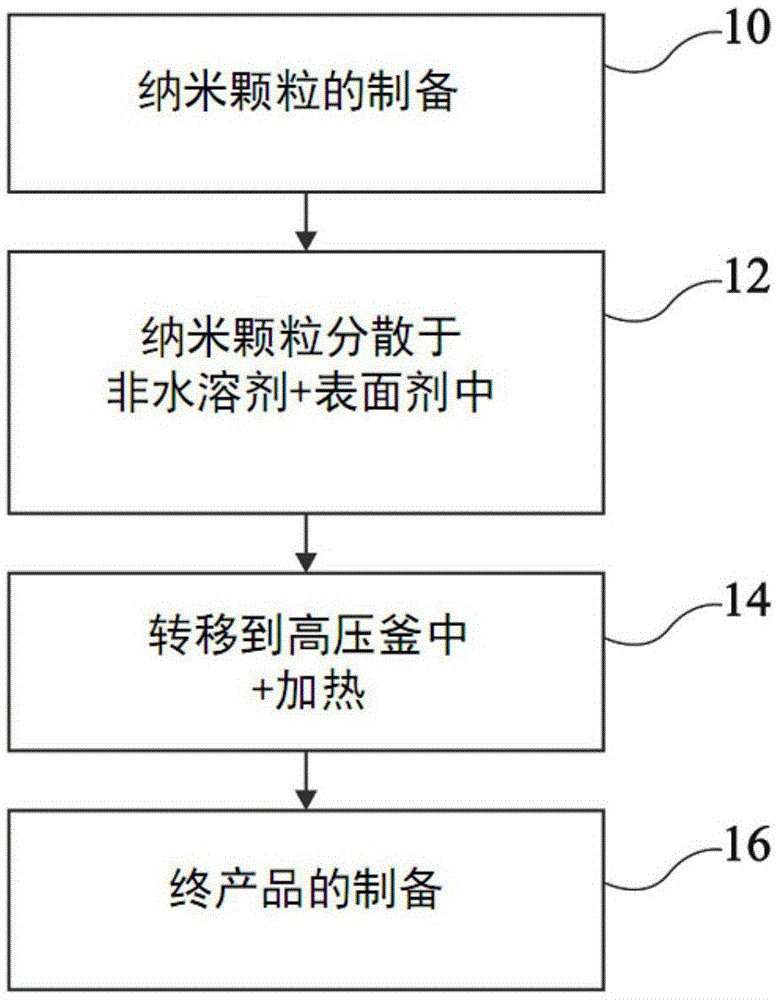
图1以方框图的形式示出了制造光致发光材料的纳米颗粒的方法的一个实施方案。该方法包括相继的步骤10至16。
在步骤10中,制造光致发光材料的纳米颗粒。
根据一个实施方案,所述光致发光材料是铝酸盐、硅酸盐、氮化物、氮氧化物、氟化物或硫化物。作为一个实例,所述光致发光材料能够在波长在250nm至500nm,优选360nm至480nm范围内的光激发下发射波长为400nm至700nm的光。
根据一个实施方案,所述光致发光材料主要包含铝酸盐,特别是根据下式(1)的钇铝石榴石:
(y(1-x)r1x)3(al(1-y)r2y)5o12(1)
或者根据下式(2)的镥铝石榴石:
(lu(1-x)r1x)3(al(1-y)r2y)5o12(2)
其中r1和r2独立地选自包括稀土、碱土金属和过渡金属的元素,并且x和y各自独立地在0至1之间变化。优选地,r1和r2独立地选自铈、钐、钆、硅、钡、铽、锶、铬、镨和镓。
作为在所需波长范围内吸收和发射光的氮化物的一个实例,可提及以下:caalsin3:eu、(ca,sr)alsin3:eu、ca2si5n8:eu或(ca,sr)si5n8:eu。
作为吸收和发射所需波长的光的氟化物的一个实例,可提及式x2mf6:mn(其中x可以是k或na,m可以是si、ge、sn或ti)的氟化物。
作为在所需波长范围内吸收和发射光的硫化物的一个实例,可提及以下:cas:eu、srca:eu、(sr,ca)s:eu和srga2s4:eu。
作为在所需波长范围内吸收和发射光的铝酸盐的一个实例,可提及以下:y3al5o12:ce、(y,gd)3al5o12:ce、tb3al5o12、(y,tb)3al5o12、lu3al5o12:ce和y3(al,ga)5o12。
作为在所需波长范围内吸收和发射光的硅酸盐的一个实例,可提及以下:(sr,ba)2sio4:eu、sr2sio4:eu、ba2sio4:eu、ca2sio4:eu、ca3sio5:eu和sr3sio5:eu。
在步骤12中,形成所述光致发光颗粒在非水溶剂(即含少于0.02重量%的水)中的胶体分散体。所述非水溶剂优选为极性质子非水溶剂。根据一个实施方案,所述非水溶剂是醇,特别是选自包括甲醇、乙醇、丙醇、丁醇、戊醇、己醇和异丙醇的组。根据一个实施方案,所述胶体分散体包含10mg至100mg光致发光颗粒/ml溶剂(10mg/ml-100mg/ml)。
在步骤14中,进行所述纳米范围颗粒的官能化。为此,将所述胶体分散体置于高压釜中。
在第16步中,制备终产品。所述终产品是例如液体溶液、粘性溶液、粉末、涂层或在基底上、特别是用于发光二极管的基底上的沉积物的形式,固体膜的形式等。
图2示出了能够在步骤14中使用的高压釜20的一个实施方案。高压釜20包括待处理产品22放置在其中的腔室21。管道23能够将气体引入腔室21。管道23的开和关由闸门24控制。管道25使得能够从腔室21取样。管道25的开和关由闸门26控制。腔室21被加热器带27部分包围。压力传感器28能够测量腔室21中的压力。由马达30驱动的搅拌器29能够在腔室21中搅拌产品22。控制单元31(例如包括计算机)连接至压力传感器28、加热器带27、闸门24、26和马达30。控制单元31能够控制加热器带27和闸门24、26。控制单元31能够调节腔室21中的温度和压力并可控制搅拌器29的启动和停止。
再次考虑图1,根据一个实施方案,所述高压釜中的压力在步骤14的整个持续时间内保持基本恒定。例如,所述高压釜中的压力可以根据所用溶剂和加热温度在20bar(2mpa)至100bar(10mpa)之间变化。也可以通过向所述高压釜中引入气体(例如氮气)来调节所述压力。根据一个实施方案,所述高压釜中的温度在步骤14的整个持续时间内保持基本恒定。例如,所述高压釜中的温度为25℃至300℃,优选150℃至250℃。所述分散体中颗粒的搅拌可以在所述高压釜中进行。
根据一个实施方案,在将所述光致发光颗粒的胶体分散体置于高压釜中之前,将至少一种表面剂添加至其中。
根据一个实施方案,所述表面剂是硅烷型偶联剂。根据本说明书,表述“硅烷型偶联剂”或“硅烷偶联剂”是指能够通过化学键或物理键与有颗粒分散在其中的基质或溶剂结合并能够与颗粒表面化学结合的反应物。每种硅烷偶联剂包含能够与基质或溶剂结合的第一部分和能够与颗粒表面结合的第二部分。根据一个实施方案,所述胶体分散体包含1mg至100mg偶联剂/ml溶剂。所述硅烷偶联剂和发光纳米颗粒之间的质量比可以在5至0.01之间,优选3至0.1之间变化。所述硅烷偶联剂倾向于分布在每个光致发光颗粒的周边并形成围绕每个颗粒的可变密度层。
硅烷偶联剂是具有下列化学式(3)的有机官能化合物:
rnsix4-n(3)
其中n等于1、2或3,x表示可水解的基团,特别是烷氧基、卤素基团或胺基,并且r是不可水解的有机基团。作为一个实例,所述有机硅烷具有式rnsi(or’)4-n。
根据另一个实施方案,所述表面剂是具有以下化学式(4)的化合物:
r-y-r’(4)
y是包含选自包括碳、氢和氧的组的至少一个原子的链。y被称为间隔体。该间隔体能够在某些情况下调节光致发光颗粒的亲水/疏水特性。该间隔体可能还能够调节光致发光颗粒之间的距离。r和r’可以相同并且属于以下化学基团:硫醇(-sh)、羧酸(-cooh)、醇(-oh)、胺(-nh2)或丙烯酸酯。它们也可以是非反应性官能团,例如聚乙二醇(-peg)或芳基。r和r’也可以是单官能的(仅具有一个官能团)、双官能的(2个官能团)、三官能的(3个官能团)或四官能的(4个官能团)。
图3是根据胶体分散体的制造方法的一个实施方案在步骤14结束时获得的光致发光纳米颗粒的局部简化截面图。所述光致发光材料的每个颗粒32被偶联剂层31包围。每个颗粒32可以被所述偶联剂或多或少地完全覆盖。所述偶联剂的不能与颗粒结合的所有部分都朝向颗粒的外侧,这由图3中的附图标记34示出。
根据一个实施方案,所述硅烷偶联剂选自例如包括以下物质的组:
正丙基三甲氧基硅烷、烯丙基三甲氧基硅烷、正丙基三乙氧基硅烷、三甲氧基(7-辛烯-1-基)硅烷、三甲氧基(十八烷基)硅烷、正辛基三甲氧基硅烷、正辛基三乙氧基硅烷、甲氧基(三亚乙基氧基)丙基三甲氧基硅烷、3-氨基丙基三甲氧基硅烷、苯基三甲氧基硅烷、二甲氧基(甲基)辛基硅烷、3-巯基丙基三甲氧基硅烷、3-(甲基丙烯酰氧基)丙基三甲氧基硅烷、3-异氰酸酯基丙基三乙氧基硅烷、3-异氰酸酯基丙基三甲氧基硅烷、2-[甲氧基(聚亚乙基氧基)6-9-丙基]三甲氧基硅烷、3-环氧丙氧基丙基三甲氧基硅烷、n-(3-丙烯酰氧基-2-羟丙基)-3-氨基丙基三乙氧基硅烷和双[3-(三乙氧基甲硅烷基)丙基]脲。
根据一个实施方案,所述硅烷偶联剂可以是如氯代(二甲基)十八硅烷、氯代(十二烷基)二甲基硅烷或氯代(癸基)二甲基硅烷的碳氯代硅烷类,或如氯代二甲基(3,3,3-三氟丙基)硅烷或全氟癸基三氯硅烷的氟化氯代硅烷类。
根据一个实施方案,所述偶联剂可以进一步在颗粒表面一起反应并形成全部或部分包围每个颗粒的新化合物。
根据一个实施方案,所述有机硅烷在非水溶剂中反应形成si-o-si键。有利地,所述有机硅烷在非水介质中的反应不会引起硅烷醇(si-oh)的形成。公认地,在光致发光颗粒的表面存在如si-oh基团的羟基型基团(-oh)对颗粒的光输出具有负面影响(特别是因为这些基团形成电荷载体的陷阱)。
作为一个例子,已知,仅根据下式(5)、(6)、(7),氯化硅(sicl4)与式ror的无水醚的反应可以得到具有si-or型表面基团的sio2颗粒:
合成:si-cl+r-o-r→si-or+r-cl(5)
经:si-or+si-cl→si-o-si+r-cl(6)
或经:si-or+si-or→si-o-si+r-o-r(7)缩合;
图4是被通过在硅烷偶联剂之间形成si-o-si键而获得的具有可变密度的硅氧烷层33包围的颗粒32的局部简化截面图。r基团朝向颗粒32的外侧。
根据一种变形,步骤14可以包括用于构成光致发光颗粒的全部或部分前体的表面处理步骤。该步骤可以在官能化步骤之前或期间进行,优选在表面官能化步骤之前进行。例如,在钇、铝、铈和氧源存在下的表面处理可用于处理所述yag:ce纳米颗粒的表面。对于ca2mgsi2o7:eu纳米颗粒,可以将硅、镁、钙、铕和氧源的全部或部分与如前所述在高压釜中加热后的纳米颗粒混合。所述前体将在分散体中一起反应并形成完全或部分包围每个颗粒的层。
图5是在已经用全部或部分用于构建光致发光颗粒的前体进行了表面处理的步骤时,在步骤14结束时获得的颗粒32的局部简化截面图。所述前体已在分散体中共同反应并已形成完全或部分包围颗粒32的层35。
根据另一种变型,可以将一种或多种光致发光物质添加到步骤14的混合物中。所述光致发光物质可以是有机的、无机的或混合型的,其发射与颗粒20相同或不同发射波长的光。在步骤14结束时,所述光致发光物质将位于由所述偶联剂形成的层内侧或该层外侧。
图6是当在步骤14中将光致发光物质添加至混合物时,在步骤14结束时获得的颗粒32的局部简化截面图。光致发光物质36已被捕获在层33中。
根据一个实施方案,所述光致发光物质可以是量子点。所述量子点可以通过共价化学键结合至由所述偶联剂形成的层。例如,3-巯基丙基三甲氧基硅烷可用作偶联剂。三甲氧基硅烷基团将与所述光致发光颗粒的表面形成共价键,而朝向所述颗粒外侧的硫醇(-sh)基团将与所述量子点的表面形成共价键。
图7是在步骤14结束时获得的颗粒32的局部简化截面图,其中量子点37结合至层33的表面。
步骤14的持续时间可以为30分钟至多天,优选30分钟至48小时,优选10小时至20小时。加热时间特别取决于加热温度、溶剂和所用的表面剂。
根据一个实施方案,官能化步骤14之前可以是对步骤12中获得的纳米颗粒进行表面处理的附加步骤。该步骤可以包括:在室温下将纳米颗粒分散体与二氧化硅前体混合从1小时至24小时的持续时间。所述二氧化硅前体例如是teos。然后,例如,可以在氨(nh4oh)存在下根据
步骤16包括制备适合于目标应用的终产品。步骤16可以包括:回收在步骤14结束时获得的胶体分散体中的颗粒。该回收步骤可以包括纳米颗粒沉淀的步骤,例如通过添加所述纳米颗粒的反溶剂。“反溶剂”是指对所述纳米颗粒没有“化学”亲和力的任何溶剂。所述反溶剂的选择将取决于所述纳米颗粒的性质和表面化学。所述反溶剂通常是非质子极性溶剂,例如丙酮,或质子极性溶剂,例如乙醇。作为一种变型,可以通过离心回收含有纳米颗粒的固相。步骤16之后可以进行颗粒纯化步骤,特别是除去未反应的前体和寄生反应产物以及步骤14所用的溶剂。然后将纳米颗粒干燥,例如在25℃至80℃范围的温度下干燥1小时至12小时的时间。于是获得纳米颗粒粉末。
可以将在步骤16中获得的纳米颗粒粉末添加至可选地还包含粘合剂(例如树脂)的溶剂,以形成光致发光颗粒的流体或粘性组合物。可以选择与所述光致发光颗粒结合的表面剂以改善所述光致发光颗粒与所述溶剂和/或粘合剂之间的相容性,例如增加所述光致发光颗粒在所述溶剂和/或粘合剂中的分散。
所述光致发光组合物的施用方法,特别是形成涂层,可以对应于所谓的添加法,例如通过在所需位置直接印刷所述光致发光组合物,例如通过喷墨印刷、照相凹版印刷、丝网印刷、柔性版印刷、喷涂、滴涂或通过将基质浸入所述光致发光纳米颗粒溶液(浸涂)。
图8以方框图的形式示出了制造光致发光材料的纳米颗粒的前述步骤10的一个实施方案。该方法包括相继的步骤101和102。
在步骤101中,通过制造光致发光材料颗粒的已知方法形成平均尺寸大于1μm的颗粒。作为一个实例,所述方法包括固态反应。作为一个实例,将固体前体即粉末形式的光致发光材料的组分混合、研磨并在高温下加热,例如加热至高于1600℃的温度,以形成具有所需组成和晶体相的颗粒粉末。可以将所述粉末在还原气氛,例如氢气(h2)下进行退火。
在步骤101结束时,光致发光颗粒的平均尺寸为大于1μm,可以为10μm至15μm。
在步骤102中,通过例如使用球磨机将颗粒进行研磨来降低在步骤101中获得的光致发光颗粒的平均尺寸。所述研磨优选为湿磨,其中所述颗粒分散在溶剂中。所述溶剂优选为非水溶剂,特别是极性质子非水溶剂。根据一个实施方案,所述非水溶剂是醇,优选选自包括甲醇、乙醇、丙醇、丁醇、戊醇、己醇和异丙醇的组,特别是乙醇。在步骤102结束时,所述光致发光颗粒的平均尺寸小于1μm,例如在100nm至500nm的范围内。
步骤102使用的溶剂可以与之后在步骤12使用的溶剂相同。当步骤12使用的溶剂与步骤102使用的溶剂相同时,步骤12使用的光致发光纳米颗粒的胶体分散体可以对应于步骤102结束时得到的分散体。然而,在步骤12中使用与步骤102使用的溶剂不同的溶剂可能是有利的。实际上,可能期望在步骤102中使用改善研磨操作性能的第一溶剂,例如具有低粘度的溶剂,并在步骤14中使用更适合于所实施的处理的第二溶剂,例如沸点高于步骤102使用的溶剂的溶剂。
根据另一个实施方案,在制备光致发光材料的纳米颗粒的步骤10中实施的方法直接得到纳米范围颗粒,而基本上不形成平均尺寸大于一微米的颗粒。作为一个实例,所述纳米颗粒形成方法是水热法。根据一个实施方案,所述纳米颗粒是量子点。
所述量子点是纯的半导体纳米晶体(si、ge)或ii-vi型化合物(cds、cdse、cdte、zno、znse、zns)、iii-v型化合物(gaas、inp、inas、gan)、iv-vi型化合物(pbs、pbse、pbte)、i-vii型化合物(cucl),v-vi型化合物(bi2te3)或ii-v型化合物(cd3as、zn3p2、zn3as2),其直径通常为2nm至10nm。作为“量子限制”现象的直接结果,这种材料具有通过控制其尺寸可调节的荧光性质。在过去的十五年中已经开发了两种主要的量子点胶体合成方法:所谓的“金属-有机”和“水热”合成。金属有机合成基于将前体a在高温(270℃-300℃)下和前体b(例如,在cdse的情形中,a对应于se2-且b对应于cd2+)快速注入配位溶剂,如三辛基氧化膦(topo)、十六烷基胺(hda)、油胺,或非配位溶剂,如1-十八烯(ode)。水热合成在水性介质中进行且通常在室温下进行前体的注入。然后将混合物回流(100℃)或在高压釜中加热至高于150℃的温度。最常用于制造量子点并能够实现最高光输出的合成是金属-有机型方法。然而,该方法产生仅与非极性有机溶剂(甲苯、氯仿、己烷等)混溶的疏水性颗粒。不幸的是,大多数这些溶剂对人类和环境有毒。为了使颗粒与其他极性溶剂(低毒或无毒)混溶,配体交换是必要的。应注意,表面配体交换很经常地伴随着量子点的光致发光量子效率的降低。类似地,由量子点的合成产生的疏水配体(通过金属-有机方法)与用于使颗粒成形的所有类型的包封剂不相容,例如有机硅树脂。将所述疏水量子点与有机硅树脂混合时,可以观察到剧烈的颗粒团聚和絮凝。此外,由于表面金属的氧化,量子点通常随时间而具有差的稳定性,这导致光输出随时间的显著降低。因此,期望开发一种量子点表面处理的通用方法,该方法既能够保护量子点免受氧化又能够为它们提供特定表面功能,所述特定表面功能使其能够根据所需的应用和成形与其分散介质(包封剂、溶剂、配体、含有树脂的组合物等)混溶,而不因此导致原始颗粒(在表面处理之前)的光输出的损失。
在所述纳米颗粒是量子点的情况下,除有机硅烷之外,可以使用其他特定偶联剂。根据一个实施方案,这些特定偶联剂可具有至少两个硫醇型官能团(-sh)。可以使用以下偶联剂:1,6-己烷二硫醇(其具有两个硫醇官能团)、三羟甲基丙烷三(3-巯基丙酸酯)(其具有三个硫醇官能团)和季戊四醇四(3-巯基丙酸酯)(其具有四个硫醇官能团)。至少一个所述硫醇官能团在纳米颗粒的表面发生反应,而剩余的硫醇官能团朝向颗粒的外侧并可与特定的单体反应以在量子点周围形成有机层。根据一个实施方案,所述特定单体应具有至少两个丙烯酸官能团(ch2=chcoo-)并且可以例如是:聚(乙二醇)二丙烯酸酯(其具有两个丙烯酸官能团)、季戊四醇三丙烯酸酯(其具有三个丙烯酸官能团)、季戊四醇四丙烯酸酯(其具有四个丙烯酸官能团)。通过加成反应进行所述硫醇官能团和所述丙烯酸官能团之间的偶联,并产生围绕纳米颗粒的由丙烯酸酯官能团封端的有机配体链。所述加成反应可以通过胺或通过自由基引发剂来催化。
图9示出了在使用季戊四醇四(3-巯基丙酸酯)作为特定配体并使用季戊四醇四丙烯酸酯作为特定单体的情况下的该实施方案。然后,在纳米颗粒表面产生的丙烯酸酯官能团可以用作交联基质,以将量子点包封在聚合物(例如pmma)或树脂(例如硅树脂)或抗蚀剂中。
抗蚀剂可以是环氧树脂、丙烯酸酯、硅树脂等类型,优选丙烯酸酯型。根据一个实施方案,所述丙烯酸酯抗蚀剂包含至少以下5种组分:
至少一种丙烯酸类基础单体或低聚物,其形成抗蚀剂的基底;
粘合剂,其用于改善抗蚀剂的机械性能并使抗蚀剂在照射后易于显影;
光引发剂,其用于引发抗蚀剂交联反应;
至少一种类型的散射颗粒,其用于提高量子点/抗蚀剂层的光提取效率;
至少一种溶剂,其用于将混合物置于溶液中。所述溶剂还能够调节混合物的粘度并使其适应成型方法的规格。
所述丙烯酸类基础单体或低聚物可选自包括聚乙二醇二丙烯酸酯、三丙二醇二丙烯酸酯、3-甲基-1,5-戊二醇二丙烯酸酯、二丙二醇二丙烯酸酯、二丙烯酸己二醇酯、三羟甲基丙烷三丙烯酸酯、三羟甲基丙烷、三丙烯酸酯、三羟甲基丙烷三丙烯酸酯、季戊四醇三和四丙烯酸酯、双三羟甲基丙烷四丙烯酸酯、二季戊四醇六丙烯酸酯和二季戊四醇六丙烯酸酯的组。
所述粘合剂可以是分子量为1000至40000g/mol、优选2000至20000g/mol的聚合物或共聚物。所述粘合剂应具有至少等于100mgkoh/g粘合剂的酸值,以允许在照射后的显影期间更好地提取抗蚀剂,并且还使抗蚀剂更好地粘合至目标基底。所述粘合剂的组成还应考虑量子点和散射颗粒的化学性质,以提供与光敏组合物更好的相容性。粘合共聚物可以例如包括以酸官能团为末端的单体(例如丙烯酸)、以醇官能团为末端的丙烯酸酯单体(例如甲基丙烯酸2-羟乙酯)和以芳族官能团为末端的丙烯酸酯单体(例如甲基丙烯酸苄酯)。以乙烯基官能团为末端的单体或不饱和聚合物也可添加至粘合剂,如苯乙烯、甲基乙烯基醚、4-甲基苯乙烯。
所述散射颗粒可以是sio2、al2o3、tio2、zro2、zno或baso4颗粒。所述散射粒径可以为0.005μm至10μm,优选0.05μm至1μm。
可以使用多种溶剂(或溶剂混合物)。根据量子点的表面基团的类型、散射颗粒以及与粘合剂、抗蚀剂等的相容性,所述溶剂可以是极性或非极性类型。
所述量子点/光敏组合物混合物可通过不同的沉积方法施加至所述基底,例如旋涂、浸涂、溅射或层涂。
本发明人已经进行了实施例。对于包括研磨步骤的实施例,例如先前结合图8描述的步骤102,通过使用光致发光颗粒的悬浮体进行所述研磨步骤。通过将10g光致发光颗粒在45ml乙醇中与240g直径为0.5mm的zro2球混合来形成悬浮体。将所述悬浮颗粒研磨30分钟,且研磨机的转速为1800转/min。
对于包括纳米颗粒官能化步骤的实施例,例如先前结合图1描述的步骤14,通过加入过量的乙腈使官能化的纳米颗粒沉淀。通过离心回收了固相,然后在25℃和80℃之间干燥12小时以形成纳米颗粒粉末。
对所制造的光致发光粉末和光致发光颗粒分散体进行内量子效率qyint、吸收系数abs和外量子效率qyext的测量。内量子效率qyint、吸收系数abs和外量子效率qyext由以下关系式(4)定义:
qyint=nem/nabs(4)
abs=nabs/nexc
qyext=qyint*abs
其中nem和nabs分别是由光致发光材料发射和吸收的光子数,nexc是激发源发射的光子总数。qyint和abs值由测量设备直接得到。由此可以从这些值推导出外量子效率qyext。
通过使用配备有积分球的hamamatsucg-2光谱仪(250-900nm)进行内量子效率qyint和吸收系数abs的测量。外量子效率qyext的值具有5%的误差容限。
在实施例1至6中,制造了yag:ce3+光致发光颗粒。当这些颗粒被蓝光激发时,它们能够发射黄光。
对比实施例1
用freeradicaltechnologyco.,ltd(台湾)市售的编号pf-y46w200的yag:ce3+微粒粉末作为对照粉末。
所述微粒是通过固态反应合成获得的。在实施例1中,没有研磨步骤,也没有通过偶联剂进行官能化的步骤。
图10示出了所述yag:ce3+微粒粉末的粒度曲线。d50为14μm,d90为20μm,d10为8μm。发射带的最大值位于558nm处。
测量了内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(i)中:
qyint(%)absqyext(%)
950.9187
表i
对比实施例2
用实施例1的yag:ce3+微粒粉末形成了纳米颗粒粉末。
进行了研磨步骤。未进行用偶联剂官能化的步骤。
图11示出了研磨后获得的yag:ce3+纳米颗粒粉末的粒度曲线。
测量了内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(ii)中:
qyint(%)absqyext(%)
680.854
表ii
通过研磨实施例1的粉末而未进行官能化步骤所获得的纳米颗粒粉末的外量子效率qyext比实施例1的粉末的量子效率低33个点以上。
对比实施例3
通过溶剂热法制造了yag:ce3+纳米颗粒的粉末。
通过将56.16mmol水合乙酸钇、0.05mmol水合乙酸铈(iii)和94.55mmol异丙醇铝在包含450ml1,4-丁二醇和60ml二乙二醇的溶剂混合物中混合,形成了胶体分散体。将该混合物在高压釜中在300℃下加热1小时。将获得的胶体分散体冷却至室温。通过添加过量的乙腈使纳米颗粒沉淀。通过离心回收固相,然后在80℃下干燥12小时以形成纳米颗粒粉末。纳米颗粒粉末在1500℃下退火4小时。在退火之前,平均纳米颗粒尺寸为40nm。在退火之后,平均纳米颗粒尺寸在100nm至900nm的范围内。
测量了内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(iii)中:
qyint(%)absqyext(%)
490.7135
表iii
实施例4
用实施例1的yag:ce3+微粒粉末形成了纳米颗粒粉末。
进行了研磨步骤。进行了官能化步骤。对于该官能化步骤,溶剂是乙醇,且偶联剂是三甲氧基十八烷基硅烷(tmods,sigmaaldrich)。将纳米颗粒浓度调节至35mg/ml乙醇。纳米颗粒与偶联剂的质量比为1:1。将分散体置于高压釜中,在150℃、20bar至30bar的压力下保持17小时。
在官能化步骤之后通过过量添加乙醇(反溶剂)回收了纳米颗粒粉末。乙醇的加入导致颗粒的非常快速的倾析。这证明在tmods存在下在高压釜中处理后颗粒表面变得疏水。实现了官能化纳米颗粒的粉末在1,2-二氯苯中的分散,1,2-二氯苯是非极性溶剂。
图12示出了1,2-二氯苯中的yag:ce3+纳米颗粒分散体的粒度曲线。在室温下数周后,所述官能化的纳米颗粒在溶剂中基本稳定。
测量了内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(iv)中:
qyint(%)absqyext(%)
770.970
表iv
相对于实施例2和3中获得的纳米颗粒粉末的外量子效率qyext,实施例4中获得的纳米颗粒粉末的外量子效率qyext增加了。
图13示出了光致发光强度pl(以任意单位)根据辐射pl的波长λ(以纳米为单位)变化的曲线c1、c2和c3,所述辐射是由分别在实施例1、2和4中制造的光致发光粉末接收波长为460nm的光辐射而发射的。所述官能化步骤能够增加在研磨步骤之后获得的纳米颗粒粉末的光致发光强度。
图14示出了对在空气中干燥12小时的光致发光纳米颗粒粉末进行的x射线衍射分析的结果。结果表明,所有观察到的衍射峰对应于y3al5o12的结晶相。未能观察到杂质或寄生相。衍射峰的加宽证实了微晶具有纳米范围的直径,通过光的动态散射以及通过电子透射显微镜测量也是如此。
实施例5
用实施例1的yag:ce3+微粒粉末形成了纳米颗粒粉末。
进行了研磨步骤。进行了官能化步骤。对于官能化步骤,溶剂是乙醇,且偶联剂是(3-环氧丙氧基丙基)三甲氧基硅烷(gptms,sigmaaldrich)。将纳米颗粒浓度调节至50mg/ml乙醇。所述纳米颗粒与所述偶联剂之间的质量比为1:1。将分散体置于高压釜中,在150℃下、20bar至30bar的压力下放置25小时。
在官能化步骤之后通过离心回收了纳米颗粒粉末。实现了官能化纳米颗粒的粉末在二乙二醇二乙醚中的分散,二乙二醇二乙醚是极性溶剂。
图15示出了在官能化步骤后获得的yag:ce3+纳米颗粒粉末的粒度曲线。该曲线接近图11的曲线。
测量了内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(v)中:
qyint(%)absqyext(%)
750.860
表v
相对于实施例2和3中获得的纳米颗粒粉末的外量子效率qyext,实施例5中获得的纳米颗粒粉末的外量子效率qyext增加了。
实施例6
用实施例1的yag:ce3+微粒粉末形成了纳米颗粒粉末。
进行了研磨步骤。进行了预处理步骤,其中在研磨步骤中获得的纳米颗粒分散体已经与teos并与含有30%氨(nh4oh)的水溶液混合。然后将该混合物在70℃下加热7小时。将光致发光纳米颗粒浓度调节至18mg/ml。氨溶液和teos之间的体积比为0.5mlteos/0.4mlnh4oh。teos完全水解/缩合后的sio2质量约等于光致发光纳米颗粒质量的5%。然后进行了官能化步骤。对于该官能化步骤,溶剂是乙醇,偶联剂是甲基丙烯酸(3-三甲氧基甲硅烷基)丙酯(tmspma,sigmaaldrich)。将光致发光纳米颗粒浓度调节至30mg/ml乙醇。纳米颗粒与偶联剂之间的质量比为1:0.5。将分散体置于高压釜中,在150℃、20bar至30bar的压力下保持17小时。
在官能化步骤之后回收了纳米颗粒粉末。实现了官能化纳米颗粒粉末在四氢呋喃(thf)中的分散,四氢呋喃是极性溶剂。所述光致发光纳米颗粒分散体随时间而保持稳定(无倾析)。
图16示出了在所述官能化步骤后获得的yag:ce3+纳米颗粒粉末的粒度曲线。该曲线接近图11的曲线。
测量了内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(vi)中:
qyint(%)absqyext(%)
750.967.5
表vi
相对于实施例2和3中获得的纳米颗粒粉末的外量子效率qyext,实施例6中获得的纳米颗粒粉末的外量子效率qyext增加了。
在以下实施例7-10中,制造了lu3al5o12:ce3+(luag:ce)光致发光颗粒。当这些颗粒被蓝光激发时,它们能够发射绿光。
对比实施例7
用由freeradicaltechnologyco.,ltd(台湾)市售的编号pf-x16w200的luag:ce3+微粒粉末作为对照粉末。
所述微粒是通过固态反应合成获得的。在实施例7中,没有研磨步骤,也没有通过偶联剂进行官能化的步骤。
图17示出了所述luag:ce3+微粒粉末的粒度曲线。d50为15μm。对于534nm的波长获得发射峰的最大值。
测量了内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(vii)中:
qyint(%)absqyext(%)
950.8884
表vii
对比实施例8
用实施例7的luag:ce3+微粒粉末形成了纳米颗粒粉末。
进行了研磨步骤。未进行用偶联剂官能化的步骤。
测量了内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(viii)中:
qyint(%)absqyext(%)
78.10.70254
表viii
通过研磨实施例8的粉末而未进行官能化步骤所获得的纳米颗粒粉末的外量子效率qyext比实施例7的粉末的量子效率低30个点以上。
实施例9
用实施例7的luyag:ce3+微粒粉末形成了纳米颗粒粉末。
进行了研磨步骤。进行了官能化步骤。对于该官能化步骤,溶剂是乙醇,且偶联剂是三甲氧基十八烷基硅烷。将光致发光纳米颗粒浓度调节至25mg/ml乙醇。纳米颗粒与偶联剂的质量比为1:1。将分散体置于高压釜中,在150℃、20bar至30bar的压力下保持20小时。
在官能化步骤之后回收了纳米颗粒粉末。进行了官能化纳米颗粒粉末在1,2-二氯苯中的分散。
图18示出了官能化步骤之后1,2-二氯苯中的luag:ce3+纳米颗粒分散体的粒度曲线。
测量了内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(ix)中:
qyint(%)absqyext(%)
76.40.82463
表ix
相对于实施例8中获得的纳米颗粒粉末的外量子效率qyext,实施例10中获得的纳米颗粒粉末的外量子效率qyext增加了。
图19示出了光致发光强度pl(以任意单位)根据波长λ(以纳米为单位)变化的曲线c4、c5和c6,其对应于在460nm激发下的实施例7、8和9的光致发光颗粒。所述官能化步骤能够增加在研磨步骤之后获得的纳米颗粒粉末的光致发光强度。
实施例10
首先,将yag:ce纳米颗粒用teos/nh4oh进行了处理,并通过三甲氧基(7-辛烯-1-基)硅烷(sigmaaldrich)偶联剂根据与实施例6中使用的相同的操作模式进行了官能化。洗涤并纯化后,将所述光致发光纳米颗粒再次以50mg/ml的浓度分散在1,2-二氯苯中。然后,将30ml该纳米颗粒溶液与0.1g由三甲基甲硅烷基官能团封端的甲基氢-二甲基硅氧烷聚合物(硅氢化物,ps123,unitedchemicaltechnologies)和0.02g含有0.1wt%铂二乙烯基四甲基硅氧烷(sip6830.3,gelestinc.)的溶液进行混合。将该混合物在约50℃加热10分钟。然后,将0.5g由二甲基乙烯基官能团封端的聚二甲基硅氧烷(pdms)(ps443,unitedchemicaltechnologies)添加至该第一混合物中,并将最终溶液在70℃加热10分钟至15分钟。最终的颗粒分散体在环境温度下稳定,并且可用于通过喷墨印刷沉积纳米颗粒。通过将最终溶液旋涂在玻璃板上形成了均匀的复合膜。然后将膜在90℃下加热以除去溶剂并使硅氧烷树脂交联。在该实施例中,所述光致发光纳米颗粒相对于有机硅树脂的质量百分比等于约70%。
实施例11
使用在实施例6中制备的光致发光纳米颗粒分散体。将该光致发光纳米颗粒浓度调节至20mg/mlthf。将50ml的该光致发光纳米颗粒溶液与0.9ml的甲基丙烯酸甲酯(mma,sigmaaldrich)在磁力搅拌下混合。并行地,通过将50mgaibn溶解在5mlthf中制备2,2’-偶氮双(2-甲基丙腈)(aibn,自由基聚合引发剂,sigmaaldrich)的溶液。将该aibn溶液添加至所述含有光致发光纳米颗粒的混合物中并将该最终混合物在70℃下加热4小时。获得了光致发光纳米颗粒/pmma的分散体,并可将其用于光致发光颗粒的沉积,例如通过喷墨印刷。光致发光纳米颗粒相对于pmma的质量百分比等于约60%。
在实施例12-14中,制造了(la,y)3si6n11:ce光致发光颗粒。当这些颗粒被蓝光激发时,它们能够发射黄光。
对比实施例12
用mitsubishichemicalcorporation(日本)市售的编号by-202/a的(la,y)3si6n11:ce微粒的粉末作为对照粉末。
这些微粒是通过固态反应合成获得的。在实施例12中,没有研磨步骤,也没有通过表面剂进行官能化的步骤。
图20示出了所述微粒粉末的粒度曲线,其d50为15μm。发射带的最大值位于547nm处。
测量了内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(x)中:
qyint(%)absqyext(%)
800.972
表x
对比实施例13
用实施例12的(la,y)3si6n11:ce微粒粉末形成了纳米颗粒粉末。进行了研磨步骤。未进行用表面剂官能化的步骤。
测量了内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(xi)中:
qyint(%)absqyext(%)
550.738.5
表xi
通过研磨实施例12的粉末而未进行表面修饰步骤所获得的纳米颗粒粉末的外量子效率qyext比实施例12的粉末的量子效率低33个点以上。
实施例14
用实施例12的(la,y)3si6n11:ce微粒粉末形成了纳米颗粒粉末。
进行了研磨步骤。进行了官能化步骤。在现有技术中已知:基于氮化硅的材料具有接近硅酸盐材料的表面性质。因此,纳米范围(la,y)3si6n11:ce颗粒的表面可具有例如可用于接枝硅烷型偶联剂的si-oh基团。对于所述官能化步骤,溶剂是乙醇,且偶联剂是gelest,inc市售的编号sim6492.7的2-[甲氧基(聚亚乙基氧基)6-9-丙基]三甲氧基硅烷。将纳米颗粒浓度调节至20mg/ml乙醇。纳米颗粒与偶联剂之间的质量比为1:2.5。将分散体置于高压釜中,在150℃、20bar(2mpa)至30bar(3mpa)的压力下保持17小时。
在高压釜中所述处理后,用二甘醇二乙醚溶剂交换乙醇溶剂。图21示出了(la,y)3si6n11:ce纳米颗粒在二甘醇二乙醚溶剂中的悬浮液的粒度曲线。平均粒径为256nm。在室温下数周后,所述官能化的纳米颗粒在溶剂中基本稳定。
测量了二甘醇二乙醚溶剂中所述纳米范围(la,y)3si6n11:ce颗粒的内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(xii)中:
qyint(%)absqyext(%)
700.963
表xii
相对于实施例13中获得的纳米颗粒粉末的外量子效率qyext,实施例14中获得的纳米颗粒粉末的外量子效率qyext增加了。
图22示出了光致发光强度pl根据波长λ(以纳米为单位)变化的曲线c7、c8和c9,所述波长λ是由分别在实施例12、13和14中制造的光致发光颗粒接收波长为460nm的光辐射而发射的。所述表面修饰步骤能够增加在研磨步骤之后获得的纳米颗粒粉末的光致发光强度。
获得了纳米范围(la,y)3si6n11:ce颗粒在二甘醇二乙醚溶剂中的稳定悬浮液。由此制备的颗粒悬浮液可以与丙烯酸酯型聚合物(例如pmma)混合,以形成备用于例如数字喷墨印刷的稳定磷光体油墨。也可以使用其他沉积方法,特别是气溶胶喷射印刷。(la,y)3si6n11:ce纳米颗粒/pmma混合物的粘度可在2cps(2mpa.s)和100cps(100mpa.s)之间,优选4cps(4mpa.s)和20cps(20mpa.s)之间调节。
在实施例15-18中,制造了caalsin3:eu光致发光颗粒。当这些颗粒被蓝光激发时,它们能够发射红光。
对比实施例15
用mitsubishichemicalcorporation(日本)市售的编号br-101/j的caalsin3:eu微粒的粉末作为对照粉末。
这些微粒是通过固态反应合成获得的。在实施例15中,没有研磨步骤,也没有通过表面剂进行官能化的步骤。
图23示出了所述微粒粉末的粒度曲线,其d50为16μm。发射带的最大值位于648nm处。
测量了内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(xiii)中:
qyint(%)absqyext(%)
920.8477
表xiii
对比实施例16
用实施例15的caalsin3:eu微粒粉末形成了纳米颗粒粉末。进行了研磨步骤。未进行用偶联剂官能化的步骤。
测量了内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(xiv)中:
qyint(%)absqyext(%)
500.6934.5
表xiv
通过研磨实施例15的粉末而未进行表面修饰步骤所获得的纳米颗粒粉末的外量子效率qyext比实施例15的粉末的量子效率低42个点以上。
实施例17
用实施例15的caalsin3:eu微粒粉末形成了纳米颗粒粉末。
进行了研磨步骤。进行了官能化步骤。对于该官能化步骤,溶剂是乙醇,且偶联剂是gelest,inc市售的编号sia0540.0的烯丙基三甲氧基甲硅烷。将纳米颗粒浓度调节至15mg/ml乙醇。纳米颗粒与偶联剂的质量比为1:2。将分散体置于高压釜中,在170℃、20bar至30bar的压力下保持30小时。
在官能化步骤之后通过过量添加乙醇(反溶剂)回收了纳米颗粒粉末。乙醇的加入导致颗粒的非常快速的倾析。这证明在烯丙基三甲氧基甲硅烷存在下在高压釜中处理后颗粒表面变得疏水。进行了官能化纳米颗粒粉末在1,2-二氯苯中的分散。
图24示出了1,2-二氯苯中的caalsin3:eu纳米颗粒分散体的粒度曲线。其平均粒径是350nm。在室温下数天后,所述官能化的纳米颗粒在溶剂中基本稳定。
测量了1,2-二氯苯溶剂中的纳米范围caalsin3:eu颗粒的内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(xv)中:
qyint(%)absqyext(%)
750.860
表xv
相对于实施例16中获得的纳米颗粒悬浮液的外量子效率qyext,实施例17中获得的纳米颗粒悬浮液的外量子效率qyext增加了。
图25示出了光致发光强度pl根据波长λ(以纳米为单位)变化的曲线c10、c11和c12,所述波长λ是由分别在实施例15、16和17中制造的光致发光颗粒接收波长为460nm的光辐射而发射的。所述表面修饰步骤能够增加在研磨步骤之后获得的纳米颗粒粉末的光致发光强度。
实施例18
制备了caalsin3:eu/硅氧烷基油墨。为此,将2.35g实施例17中获得的纳米范围caalsin3:eu颗粒分散在20ml1,2-二氯苯溶剂中。然后,将2.1g以两个氢化物官能团封端的聚二甲基硅氧烷(pdms)(由gelest,inc市售的编号dms-h21)和0.05gpt基催化剂(由gelest,inc市售的编号sip6832.2)添加至纳米范围caalsin3:eu颗粒的悬浮液中。然后将该混合物在90℃下加热12小时。冷却后,将0.25g由两个乙烯基官能团封端的pdms(gelest,inc市售的编号dms-v31)添加至该混合物中,然后在90℃下加热30分钟。由此获得了稳定的硅氧烷基caalsin3:eu磷光体油墨,并备用于例如喷墨印刷。
在实施例19至21中,制造了光致发光k2sif6:mn颗粒。当这些颗粒被蓝光激发时,它们能够发射红光。
对比实施例19
用mitsubishichemicalcorporation(日本)市售的编号br-301/c的k2sif6:mn微粒粉末作为对照粉末。
这些微粒是通过固态反应合成获得的。在实施例19中,没有研磨步骤,也没有通过表面剂进行官能化的步骤。
图26示出了所述微粒粉末的粒度曲线,其d50为42μm。发射带的最大值位于632nm处。
测量了内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(xvi)中:
qyint(%)absqyext(%)
910.7265.5
表xvi
对比实施例20
用实施例19的k2sif6:mn微粒粉末形成了纳米颗粒粉末。进行了研磨步骤。未进行用偶联剂官能化的步骤。
测量了内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(xvii)中:
qyint(%)absqyext(%)
350.7225
表xvii
通过研磨实施例20的粉末而未进行表面修饰步骤所获得的纳米颗粒粉末的外量子效率qyext比实施例19的粉末的量子效率低40个点以上。
实施例21
用实施例19的k2sif6:mn微粒粉末形成了纳米颗粒粉末。
进行了研磨步骤。进行了官能化步骤。在官能化步骤之前进行表面处理,其目的在于修复由研磨产生的缺陷或部分表面缺陷。通常,将20mg由sigmaaldrich市售的氟化氢钾和5mg纳米范围的二氧化硅颗粒sio2(具有12nm的平均尺寸,sigmaaldrich市售)添加至200ml的实施例20的k2sif6:mn纳米颗粒的悬浮液中,将k2sif6:mn颗粒浓度调节至10mg/ml乙醇。然后将该混合物转移到高压釜中并在170℃下加热24小时。然后,将140μl油酸添加至该混合物中,然后将其在高压釜中再次在110℃、20bar至30bar之间的压力下加热2小时。
在官能化步骤之后通过离心回收了纳米颗粒粉末。在1,2-二氯苯中进行了所述官能化纳米颗粒粉末的分散。
图27显示了在1,2-二氯苯溶剂中的k2sif6:mn纳米颗粒悬浮液的粒度曲线。其平均粒径为295nm。在室温下数天后,所述官能化的纳米颗粒在溶剂中基本稳定。
测量了1,2-二氯苯溶剂中的纳米范围k2sif6:mn颗粒的内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(xviii)中:
qyint(%)absqyext(%)
730.858.4
表xviii
相对于实施例20中获得的纳米颗粒悬浮液的外量子效率qyext,实施例21中获得的纳米颗粒悬浮液的外量子效率qyext显著增加了。
图28示出了光致发光强度pl根据波长λ(以纳米为单位)变化的曲线c13、c14和c15,所述波长λ是由分别在实施例1、2和3中制造的光致发光颗粒接收波长为460nm的光辐射而发射的。所述表面修饰和官能化步骤能够增加在研磨步骤之后获得的纳米颗粒粉末的光致发光强度。
实施例22
用najingtechnology(中国)市售的cdse/zns型量子点在甲苯溶剂中的悬浮液作为对照溶液。所述量子点由油酸配体来稳定。
这些半导体纳米晶体是通过在溶液中反应合成获得的。在实施例22中,未进行颗粒表面的修饰。
图29示出了甲苯中纳米晶体尺寸分布的曲线。应该指出,所测量的尺寸代表流体动力学半径而不是硬质颗粒的半径。所述流体动力学半径有效地对应于硬球的半径加上在颗粒周围形成的溶剂化层的厚度。所述溶剂化层含有配体和溶剂两者。平均流体动力学半径约为10nm。发射带的最大值在640nm处。图30示出了根据波长λ(以纳米为单位)的光致发光强度pl,所述波长λ是由实施例22的量子点接收波长为460nm的光辐射而发射的。
测量了内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(xix)中:
qyint(%)absqyext(%)
800.9878.5
表xix
实施例23
将实施例22的量子点溶液用于在高压釜中进行表面官能化步骤。该表面官能化分两步进行。首先,通过3-巯基丙基三甲氧基硅烷偶联剂对量子点进行表面官能化。例如,制备200ml浓度为1mg/ml的量子点溶液,并将其转移到体积为200ml的高压釜中。通过用氮气n2对该溶液进行鼓泡直至其饱和来将氧气从该量子点溶液中除去。然后将该量子点溶液置于手套箱中,并将170mg的3-巯基丙基三甲氧基硅烷偶联剂添加至该量子点溶液。然后将高压釜关严,从手套箱中取出,并在170℃下在搅拌下加热12小时。然后通过离心回收量子点,再用过量的乙醇进行清洗。重复在3-巯基丙基三甲氧基硅烷偶联剂存在下在高压釜中的处理,直到量子点在乙醇中形成稳定的悬浮液。在第二步中,将第一步产生的量子点分散在200ml无水乙醇中并转移到高压釜中。也进行了用氮气n2鼓泡直至达到饱和。将高压釜置于手套箱中,并将250mg偶联剂(3-三甲氧基甲硅烷基)丙基甲基丙烯酸酯添加至该量子点溶液中。将高压釜从手套箱中取出并在170℃下在搅拌下加热12小时。用丙二醇单甲醚乙酸酯(pgmea)对乙醇溶剂进行了交换。由此获得由甲基丙烯酸基团官能化的量子点。
图31示出了pgmea中的纳米晶体的尺寸分布曲线。其平均流体动力学半径为约66nm。该平均流体动力学半径相对于实施例22的量子点增加了。
图32示出了根据波长λ(以纳米为单位)的光致发光强度pf,所述波长λ是由实施例23的量子点接收波长为460nm的光辐射而发射的。发射带的最大值位于655nm处。
测量了内量子效率qyint、吸收系数abs和外量子效率qyext。获得的结果集于下列表(xx)中:
qyint(%)absqyext(%)
700.9868.6
表xx
实施例24
形成了量子点/光敏组合物混合物。
用实施例23的量子点溶液形成了所述量子点/光敏组合物混合物。表(xxi)示出具有各组分的质量百分比和组成的配方的一个实例。
表xxi
实施例23的粘合剂可以例如根据参考文献“journalofappliedpolymerscience,vol.109,467–474(2008).”中记载的方案来制造。量子点与表(xxi)的光敏组合物的混合在300转/分钟的恒速搅拌下远离日光进行。所述混合也可以通过行星式混合器进行。由于其表面甲基丙烯酸酯官能团,量子点对光敏组合物具有非常好的亲合力。因此,实施例23的量子点在光敏混合物中形成了稳定的悬浮液。此外,位于量子点表面的可交联甲基丙烯酸酯官能团在uv交联后使得量子点在树脂中更好的分散。
前面的实施例表明,无论光致发光纳米颗粒的化学性质如何,通过用适合的表面剂进行表面处理,纳米颗粒悬浮液的外量子效率qyext都显著增加。
- 还没有人留言评论。精彩留言会获得点赞!