一种红色荧光碳点的合成方法及应用与流程
本发明属于纳米技术领域,涉及一种碳纳米材料的制备方法,特别涉及一种红色荧光碳点的合成方法及应用。
背景技术:
碳点(c-dots)通常是尺寸为1-10nm之间的荧光碳纳米颗粒。2004年,xu等人利用凝胶电泳纯化分离电弧放电烟灰制备的单壁碳纳米管时,首次发现了这种荧光碳纳米颗粒。此后,c-dots由于其超小的粒径、优异的荧光性质、制备成本低廉、合成原料丰富易得、合成方法简单和易于功能化修饰等特点,成为一种明星材料。它们可以通过电弧放电、激光烧蚀、电化学合成、酸氧化、微波辅助合成、热解/溶剂热法、水热法等方法制备得到。与半导体量子点和有机染料相比,c-dots具有抗光漂白、抗光闪烁、水溶性好、无毒副作用和容易制备等特点,使其在生物医学应用领域有着独特的优势,被广泛地应用于生物成像、生物传感、药物运输等方面,也引起了广大研究者的兴趣。然而,大多数报道合成的c-dots为蓝色和绿色荧光。在进行生物成像时,生物样品在短波长区域具有较强的自发荧光和背景光散射,而长波长荧光可以穿透到生物样品的更深部位,使其具有更高的成像对比度。因此,需要开发长波长发射的荧光c-dots,提高其用于生物成像时的实际可操作性。专利cn201711399203.2公开了一种红色荧光碳点粉末的制备方法,以1,2,4-苯三胺和聚乙二醇为原料,通过加热反应获得红色的碳点溶液,将该碳点溶液与硅胶粉混合并在旋蒸仪上旋干即可获得红色荧光碳点粉末。该方案的优点是可有效克服碳点荧光淬灭的问题。其发射波长为400nm至520nm,且该碳点的波长随着发射波长的不同而变化,不适合用于生物成像中。
技术实现要素:
本发明提出一种红色荧光碳点的合成方法及应用,解决现有红色荧光碳点在成像时随着发射波长的变化会发生变化、且荧光性不稳定的技术问题。
本发明的技术方案是这样实现的:
一种红色荧光碳点的合成方法,步骤为:向碳源中加入剥离剂,混匀后于一定条件下反应,氧化剥离碳源,即得红色荧光碳点。
所述碳源为聚噻吩,剥离剂为浓硝酸。
所述碳源的质量(mg)与剥离剂的体积(ml)之比为20:(2-15)。
反应的条件为将温度加热至40-100℃,冷凝回流20min-24h。
所述的红色荧光碳点的合成方法,步骤为:向聚噻吩中加入浓硝酸,混匀后,升温至40-100℃,冷凝回流20min-24h,得红色荧光碳点悬浊液,经纯化得红色荧光碳点。
每mg聚噻吩中加入0.1-0.75ml的浓硝酸。
所述纯化步骤为:向制得的红色荧光碳点悬浊液中加水清洗,搅拌后静置,然后离心收集沉淀,向沉淀中加入0.1m的naoh溶液使其ph值为中性,再次离心收集沉淀,然后再将沉淀分散至体积比为1:4的n,n-二甲基甲酰胺和水的混合溶液中,透析,过硅胶柱层析色谱进行分离。
所合成的红色荧光碳点在制备生物成像试剂中的应用。
本发明具有以下有益效果:
(1)本发明提供了一种简单快速合成红色荧光碳点的方法,即使用硝酸剥离pth来制备红色荧光碳点。关于红色荧光碳点的合成案例还很少,传统的红色荧光碳点的合成主要为水热法或后期高分子包裹的方法。水热法能耗高,制备红色荧光碳点时,能选取的碳源种类十分有限。而高分子包裹的方法则步骤繁杂,且需要多次蒸干。而本发明的合成方法则是利用硝酸直接氧化剥离pth,在较低的温度下反应一段时间,即可得到红色荧光碳点。该方法操作简单,制备快速,20min内即可得到红色荧光碳点。
(2)本发明的合成方法的反应物十分简单,只需要pth和硝酸即可,合成方法的反应温度极为温和,只需要简单的加热装置和冷凝回流装置即可实现红色荧光碳点的制备,可以大大降低碳点合成的成本。
(3)本发明的合成方法的反应时间极短,20min即可制得红色荧光碳点,合成方法制备的红色荧光碳点的荧光发射峰不随激发波长的改变而改变,区别于荧光发射峰随激发波长改变而改变的碳点。
(4)本发明方法成功的利用硝酸的氧化剪切作用,将pth的π电子共轭体系断裂一部分,同时通过硝酸的氧化作用,制备红色荧光碳点,制得的碳点荧光发射波长可在575-595nm之间有效的调节。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
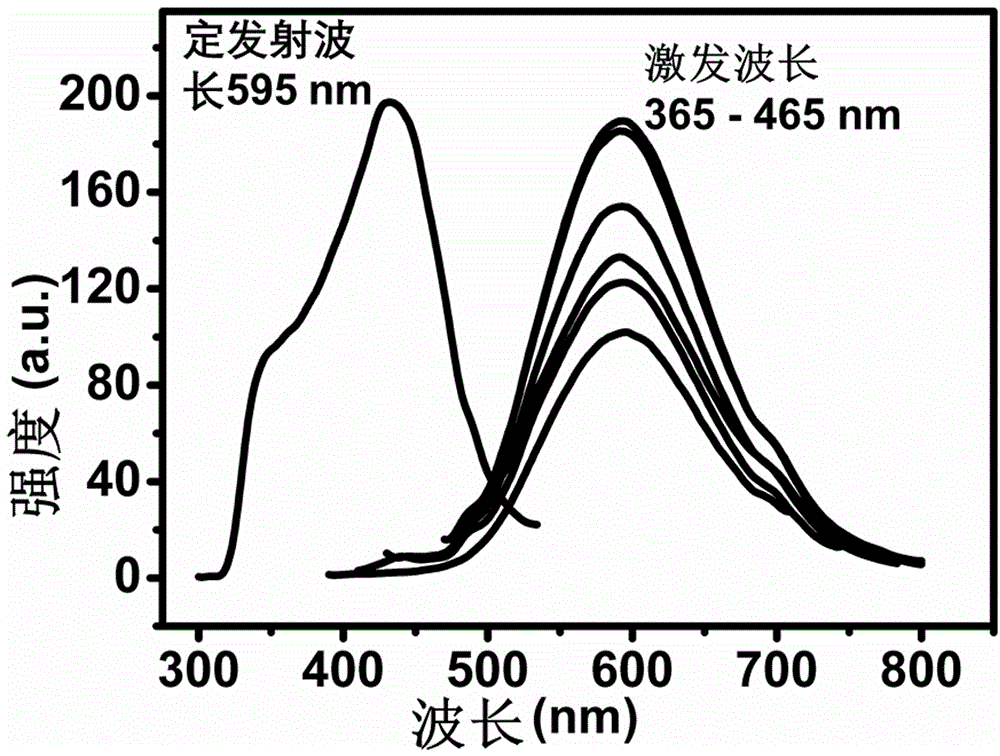
图1是实施例1碳点的荧光光谱图。
图2是实施例2碳点的荧光光谱图。
图3是实施例3碳点的荧光光谱图。
图4是实施例4碳点的荧光光谱图。
图5是实施例5碳点的荧光光谱图。
图6是实施效果例碳点应用于细胞成像图。
具体实施方式
下面将结合本发明实施例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有付出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
本实施例的一种红色荧光碳点的合成方法,步骤为:
将20mgpth和15ml浓硝酸混合均匀,在40℃下搅拌反应,冷凝回流24h,得到棕褐色的碳点溶液,该碳点的荧光发射峰位于595nm,荧光激发峰位于430nm处。
选取激发波长分别为365nm,385nm,405nm,425nm,445nm,465nm激发,结果见图1,碳点的荧光发射峰的位置不随激发波长的改变而改变,这是因为碳点内部的碳核结构相对完整的原因。
实施例2
本实施例的一种红色荧光碳点的合成方法,步骤为:
将20mgpth和15ml浓硝酸混合均匀,在60℃下搅拌反应,冷凝回流24h,得到棕红色的碳点溶液,该碳点的荧光发射峰位于590nm。
每隔一段时间取样,进行光谱测定,观测反应进程结果。选取反应至20min,40min,1h,2h,4h,8h,12h,24h的样品测定其荧光光谱,见图2,随着反应时间的推移,碳点的荧光发射峰不会发生位移,但是其荧光强度会相应增加。这是由于随着反应时间的增加,反应越完全,但是温度相对较低,不足以对碳点的结构造成大的改变。
实施例3
本实施例的一种红色荧光碳点的合成方法,步骤为:
将20mgpth和15ml浓硝酸混合均匀,在80℃下搅拌反应,冷凝回流8h,得到棕黄色的碳点溶液,该碳点的荧光发射峰位于585nm。
每隔一段时间取样,进行光谱测定,观测反应进程。选取反应至20min,40min,1h,2h,4h,8h,12h,24h的样品测定其荧光光谱,见图3,随着反应时间的推移,碳点的荧光发射峰不会发生位移,但是荧光强度会增加,最终趋于稳定。这是由于随着反应温度的增加,反应速率有所提升,但碳点的结构仍未发生大的改变。
实施例4
本实施例的一种红色荧光碳点的合成方法,步骤为:
将20mgpth震荡分散于2ml水中,加入15ml浓硝酸,将其搅拌混合均匀,在100℃条件下搅拌反应,每隔一段时间取样,进行光谱测定,观测反应进程。
选取反应至20min,40min,1h,2h,4h,6h,8h,12h,24h,48h的样品测定其荧光光谱,见图4,随着反应时间的延长,所制备碳点的荧光发射峰逐渐由595nm处蓝移到575nm,这是由于100℃条件下,硝酸的剥离作用更加强烈,随着反应的进行,碳点的尺寸逐渐变小的原因。
实施例5
本实施例的一种红色荧光碳点的合成方法,步骤为:
将20mgpth震荡分散于15ml浓硝酸,分别加入0ml,2ml,5ml,8ml水,将其搅拌混合均匀,在100℃条件下搅拌反应24h,得到碳点的溶液,测定其荧光光谱,结果见图5。随着水加入的体积越多,也即硝酸的浓度越低,得到的碳点的荧光发射峰逐渐由575nm红移到595nm。这是由于硝酸的剥离作用随着其浓度的降低而降低,得到的碳点的π电子共轭逐渐变大的原因。
实施效果例
向实施例1制备的碳点溶液中加水清洗,搅拌后静置,然后离心收集沉淀,向沉淀中加入0.1m的naoh溶液使其ph值为中性,再次离心收集沉淀,然后再将沉淀分散至体积比为1:4的n,n-二甲基甲酰胺和水的混合溶液中,透析,过硅胶柱层析色谱进行分离,得红色荧光碳点。
经过纯化的碳点应用于细胞成像,步骤为:
将人宫颈癌细胞接种于培养皿中,在37℃条件下培养,使其贴壁。用新鲜培养基将碳点稀释至50µg/ml,更换含有碳点的培养基继续培养16h,然后用无菌磷酸盐缓冲液清洗3次,用激光共聚焦显微镜观察碳点对人宫颈癌细胞的成像,结果如图6所示,与现有的细胞成像相比,本申请的碳点的荧光发射波长更长,细胞自身的荧光和背景散射光对实施例碳点荧光的干扰更弱,细胞成像效果更为显著,探测深度更大,对比度更高。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!