水性粘合剂配制剂的制作方法
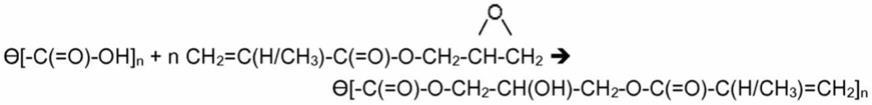
水性粘合剂配制剂
1.本发明涉及一种水性粘合剂配制剂,其包含:
2.a)至少一种有机化合物a,和
3.b)至少一种羧酸酰肼b,
4.其中所述至少一种有机化合物a
5.a1)具有至少两个任选取代的丙烯酰氧基,且
6.a2)不包含任何醛和/或酮羰基。
7.本发明还涉及一种制备所述水性配制剂的方法,以及其制备和用途。
8.当在粘合剂配制剂中使用酰肼时,可以归纳如下现有技术。
9.例如,ep
‑
a 332326公开了水性涂料体系,其粘合剂是具有羰基和酰肼基的聚氨酯聚合物。
10.根据wo 2016/000989,公开了单组分或双组分涂料组合物,其包含特定的两阶段分散聚合物和任选地还包含聚氨酯作为粘合剂。在两阶段分散聚合物具有酮基、醛基和/或乙酰乙酰氧基
‑
羰基的情况下,应指出的是,涂料组合物还可以包含交联剂,尤其是例如具有两个或更多个酰肼基的有机化合物,例如己二酸二酰肼、草酸二酰肼、邻苯二甲酸二酰肼和对苯二甲酸二酰肼。
11.因此,本发明的目的是提供一种基于酰肼化合物的替代粘合剂配制剂,其不需要包含羰基的反应组分。
12.该目的通过提供开头所定义的粘合剂配制剂实现。
13.本发明的水性粘合剂配制剂的必要成分是至少一种有机化合物a,其
14.a1)具有至少两个任选取代的丙烯酰氧基,且
15.a2)不包含任何醛和/或酮羰基。
16.根据本发明,有机化合物a是分子量≤500g/mol的分子有机化合物还是数均分子量>500g/mol的聚合有机化合物是无关紧要的,然而优选数均分子量>500g/mol的聚合有机化合物。聚合有机化合物a的数均分子量通常为>500且≤500000g/mol,有利地≥600且≤10000g/mol,特别有利地≥750且≤3000g/mol。在本发明的上下文中,数均分子量在此应通过使用所定义的聚苯乙烯标样的凝胶渗透色谱法测定。
17.重要的是有机化合物a具有至少两个任选取代的丙烯酰氧基,其中分子化合物a通常具有≥2且≤5,有利地≥2且≤4,特别有利地≥2且≤3个任选取代的丙烯酰氧基。聚合化合物a通常平均具有≥2且≤10,有利地≥2且≤5,特别有利地≥2且≤3个任选取代的丙烯酰氧基。
18.根据本发明,任选取代的丙烯酰氧基具有下式i的结构:
19.o
‑
c(=o)
‑
chr1=chr2ꢀꢀꢀ
式i
20.其中:
21.r1为氢、c1‑
c4烷基、
‑
c(=o)
‑
o
‑
c1‑
c4烷基或
‑
ch2‑
c(=o)
‑
o
‑
c1‑
c4烷基,优选为氢和甲基,特别优选为氢,且
22.r2为氢、c1‑
c4烷基、
‑
c(=o)
‑
o
‑
c1‑
c4烷基或
‑
c≡n,优选为氢。
23.在本说明书的上下文中,c1‑
c4烷基为甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基或叔丁基,其中优选甲基和乙基,特别优选甲基。
24.分子化合物a有利地为具有式ii结构的化合物:
25.r[
‑
z]
n
ꢀꢀꢀ
式ii
[0026]
其中它们通常为通式iii的分子二醇或多元醇化合物:
[0027]
r[
‑
oh]
n
ꢀꢀꢀ
式iii,
[0028]
其中oh基团已被式i的任选取代的丙烯酰氧基(z)代替,并且其中r为至少二价的非聚合有机基团,n为≥2的数。
[0029]
在这种情况下,所用的通式iii的分子二醇或多元醇化合物在结构上可以是芳族和脂族的,优选具有脂族结构的那些。
[0030]
芳族分子二醇或多元醇化合物为包含至少一个芳族环体系的那些,即纯芳族和芳脂族化合物。
[0031]
在脂族分子二醇或多元醇化合物的情况下,无环和脂环族化合物都是合适的。
[0032]
脂环族分子二醇或多元醇化合物是包含至少一个脂环族环体系的那些,而无环分子二醇或多元醇化合物仅具有直链或支化的非环状烃基。脂族分子二醇或多元醇化合物不包含任何芳族环体系。
[0033]
通式iii的分子无环二醇化合物的实例包括:乙二醇、丙烷
‑
1,2
‑
二醇、丙烷
‑
1,3
‑
二醇、丁烷
‑
1,2
‑
二醇、丁烷
‑
1,3
‑
二醇、丁烷
‑
1,4
‑
二醇、丁烷
‑
2,3
‑
二醇、戊烷
‑
1,2
‑
二醇、戊烷
‑
1,3
‑
二醇、戊烷
‑
1,4
‑
二醇、戊烷
‑
1,5
‑
二醇、戊烷
‑
2,3
‑
二醇、戊烷
‑
2,4
‑
二醇、己烷
‑
1,2
‑
二醇、己烷
‑
1,3
‑
二醇、己烷
‑
1,4
‑
二醇、己烷
‑
1,5
‑
二醇、己烷
‑
1,6
‑
二醇、己烷
‑
2,5
‑
二醇、庚烷
‑
1,2
‑
二醇、庚烷
‑
1,7
‑
二醇、辛烷
‑
1,8
‑
二醇、辛烷
‑
1,2
‑
二醇、壬烷
‑
1,9
‑
二醇、癸烷
‑
1,2
‑
二醇、癸烷
‑
1,10
‑
二醇、十二烷
‑
1,2
‑
二醇、十二烷
‑
1,12
‑
二醇、1,5
‑
己烷
‑
3,4二醇、2,2
‑
二甲基丙烷
‑
1,3
‑
二醇(新戊二醇)、2
‑
甲基戊烷
‑
2,4
‑
二醇、2,4
‑
二甲基戊烷
‑
2,4
‑
二醇、2
‑
乙基己烷
‑
1,3
‑
二醇、2,5
‑
二甲基己烷
‑
2,5
‑
二醇、2,2,4
‑
三甲基戊烷
‑
1,3
‑
二醇、频哪醇、二甘醇、三甘醇、二丙二醇和/或三丙二醇。
[0034]
优选的分子无环二醇组分为乙二醇、丙烷
‑
1,2
‑
二醇、丙烷
‑
1,3
‑
二醇、丁烷
‑
1,4
‑
二醇、戊烷
‑
1,5
‑
二醇、己烷
‑
1,6
‑
二醇和/或辛烷
‑
1,8
‑
二醇。
[0035]
脂环族二醇的实例为环戊烷
‑
1,2
‑
和
‑
1,3
‑
二醇、环己烷
‑
1,2
‑
、
‑
1,3
‑
和
‑
1,4
‑
二醇,1,1
‑
、1,2
‑
、1,3
‑
和1,4
‑
双(羟甲基)环己烷,1,1
‑
、1,2
‑
、1,3
‑
和1,4
‑
双(羟乙基)环己烷和双(4
‑
羟基环己烷)亚异丙基。
[0036]
优选环己烷
‑
1,2
‑
、
‑
1,3
‑
和
‑
1,4
‑
二醇,1,3
‑
和1,4
‑
双(羟甲基)环己烷和双(4
‑
羟基环己烷)亚异丙基。
[0037]
具有多于两个羟基的分子化合物包括,例如:三羟甲基丁烷、三羟甲基丙烷、三羟甲基乙烷、季戊四醇、甘油、双三羟甲基丙烷、二季戊四醇、双三羟甲基乙烷、山梨醇、甘露醇、双甘油、苏糖醇、赤藓醇、阿东糖醇(核糖醇)、阿糖醇(lyxitol)、木糖醇、卫矛醇(半乳糖醇)、麦芽糖醇或异麦芽酮糖醇(isomalt)。
[0038]
随后将描述由分子二醇化合物和具有多于两个羟基的分子化合物制备分子化合物a的一般方法。
[0039]
在本发明的上下文中,合适的聚合化合物a是具有至少两个任选取代的丙烯酰氧
基(z)的所有天然存在的或合成制备的聚合物。基于天然物质的聚合物a的实例是适当官能化的硝化纤维素、纤维素酯、松香、虫胶、亚麻子油和/或木油。合成制备的聚合化合物a包括例如适当官能化的缩聚产物,例如醇酸树脂、聚酯、聚醚、聚碳酸酯、聚酰胺、有机硅树脂和/或环氧树脂,以及加聚产物如聚氨酯,以及例如由聚合形式的烯属不饱和化合物组成的聚合物。这些缩聚化合物和加聚化合物通过本领域技术人员熟知的制备方法制备。
[0040]
聚合化合物a还有利地由相应的具有至少两个羟基的聚合物前体制备,例如特别是乙烯醇的均聚物或共聚物,丙烯酸2
‑
羟乙酯或甲基丙烯酸2
‑
羟乙酯的共聚物,和包含至少两个羟基的聚酯、聚醚或聚碳酸酯。
[0041]
在这种情况下,乙烯醇的均聚物或共聚物的制备是本领域技术人员所熟知的,并且通常通过金属络合物催化、阴离子催化、阳离子催化进行,特别优选通过乙烯醇的自由基催化均聚或共聚进行,或者通过乙酸乙烯酯的自由基诱导均聚或共聚且随后使乙酸酯基部分或完全水解断裂而进行。
[0042]
丙烯酸2
‑
羟乙酯或甲基丙烯酸2
‑
羟乙酯的共聚物的制备也是本领域技术人员所熟知的,并且通常通过金属络合物催化、阴离子催化、阳离子催化,特别优选通过丙烯酸2
‑
羟乙酯或甲基丙烯酸2
‑
羟乙酯与其他烯属不饱和化合物的自由基催化共聚来进行,所述烯属不饱和化合物例如为乙烯;乙烯基芳族单体如苯乙烯、α
‑
甲基苯乙烯、邻氯苯乙烯或乙烯基甲苯;乙烯基卤化物如氯乙烯或偏二氯乙烯;衍生自乙烯醇和具有1
‑
18个碳原子的单羧酸的酯如乙酸乙烯酯、丙酸乙烯酯、正丁酸乙烯酯、月桂酸乙烯酯和硬脂酸乙烯酯;衍生自优选具有3
‑
6个碳原子的α,β
‑
单烯属不饱和单羧酸和二羧酸如特别是丙烯酸、甲基丙烯酸、马来酸、富马酸和衣康酸与通常具有1
‑
12个,优选1
‑
8个,尤其是1
‑
4个碳原子的链烷醇的酯,例如特别是丙烯酸和甲基丙烯酸的甲酯、乙酯、正丁酯、异丁酯、戊酯、己酯、庚酯、辛酯、壬基、癸酯和2
‑
乙基己酯;富马酸和马来酸的二甲基或二正丁基酯;α,β
‑
单烯属不饱和羧酸的腈,例如丙烯腈、甲基丙烯腈、富马腈、马来腈;以及c4‑8‑
共轭二烯,例如1,3
‑
丁二烯和异戊二烯。
[0043]
有利地用作包含至少两个羟基的聚酯、聚醚或聚碳酸酯,所谓的聚酯醇、聚醚醇或聚碳酸酯醇的是根据din en iso 4629
‑
1测定的oh值为≥40且≤400mg koh/g聚合物,优选≥40且≤200mg koh/g聚合物,并且根据din 53240测定的酸值为<20mg koh/g聚合物的那些聚合化合物。
[0044]
可用作前体的聚醚醇有利地基本上是通式ho[ao]
m
h的聚氧化烯,其中ao为氧化乙烯、氧化丙烯、1,2
‑
环氧丁烷和/或2,3
‑
环氧丁烷,且m为≥8的数。在通过与水的酸或碱催化反应制备聚氧化烯的过程中,在每种情况下,氧化烯可以单独使用和/或作为混合物使用,其中仅由一种氧化烯形成聚亚烷基二醇,例如特别是聚乙二醇或聚丙二醇,或者由两种或更多种氧化烯形成聚亚烷基二醇,其中由此获得的聚亚烷基二醇包含统计混合物或嵌段形式的相应的两种或更多种氧化烯,这取决于制备的变型。可用作聚合物前体的聚醚醇还包括聚四亚甲基二醇,其可通过四氢呋喃的酸催化聚合得到。本领域技术人员熟知制备上述聚醚醇的相应方法。
[0045]
聚碳酸酯醇,即羟基官能化的聚碳酸酯,尤其可通过使上述分子二醇或多元醇组分,特别是脂族二醇组分,特别优选无环二醇组分与光气或碳酸二乙酯或碳酸二甲酯反应,同时除去盐酸或乙醇或甲醇而得到。
[0046]
基于乙二醇和/或新戊二醇的聚碳酸酯醇有利地以包含至少两个羟基的聚碳酸酯形式使用。
[0047]
可有利地用作羟基官能化聚合物前体的聚酯醇原则上可通过使单体或聚合二醇或多元醇与二羧酸组分反应而得到,其中基于单体脂族二醇,特别是基于单体无环二醇的聚酯醇是有利的。
[0048]
二羧酸单元可以是游离二羧酸或其衍生物。
[0049]
衍生物优选理解为意指:
[0050]
‑
单体或聚合物形式的相应酸酐,
[0051]
‑
单烷基或二烷基酯,优选单
‑
或二
‑
c1‑
c4烷基酯,特别优选单甲酯或二甲酯或相应的单乙酯或二乙酯,或
[0052]
‑
单乙烯基酯和二乙烯基酯,以及
[0053]
‑
混合酯,优选具有不同c1‑
c4烷基组分的混合酯,特别优选混合甲基乙基酯。
[0054]
分子无环二醇的实例包括:乙二醇、丙烷
‑
1,2
‑
二醇、丙烷
‑
1,3
‑
二醇、丁烷
‑
1,2
‑
二醇、丁烷
‑
1,3
‑
二醇、丁烷
‑
1,4
‑
二醇、丁烷
‑
2,3
‑
二醇、戊烷
‑
1,2
‑
二醇、戊烷
‑
1,3
‑
二醇、戊烷
‑
1,4
‑
二醇、戊烷
‑
1,5
‑
二醇、戊烷
‑
2,3
‑
二醇、戊烷
‑
2,4
‑
二醇、己烷
‑
1,2
‑
二醇、己烷
‑
1,3
‑
二醇、己烷
‑
1,4
‑
二醇、己烷
‑
1,5
‑
二醇、己烷
‑
1,6
‑
二醇、己烷
‑
2,5
‑
二醇、庚烷
‑
1,2
‑
二醇、庚烷
‑
1,7
‑
二醇、辛烷
‑
1,8
‑
二醇、辛烷
‑
1,2
‑
二醇、壬烷
‑
1,9
‑
二醇、癸烷
‑
1,2
‑
二醇、癸烷
‑
1,10
‑
二醇、十二烷
‑
1,2
‑
二醇、十二烷
‑
1,12
‑
二醇、1,5
‑
己二烯
‑
3,4
‑
二醇、2,2
‑
二甲基丙烷
‑
1,3
‑
二醇、2
‑
甲基戊烷
‑
2,4
‑
二醇、2,4
‑
二甲基戊烷
‑
2,4
‑
二醇、2
‑
乙基己烷
‑
1,3
‑
二醇、2,5
‑
二甲基己烷
‑
2,5
‑
二醇、2,2,4
‑
三甲基戊烷
‑
1,3
‑
二醇、频哪醇、二甘醇、三甘醇、二丙二醇和/或三丙二醇。
[0055]
优选使用的分子无环二醇为乙二醇、丙烷
‑
1,2
‑
二醇、丙烷
‑
1,3
‑
二醇、丁烷
‑
1,4
‑
二醇、戊烷
‑
1,5
‑
二醇、己烷
‑
1,6
‑
二醇、2,2
‑
二甲基丙烷
‑
1,3
‑
二醇和/或辛烷
‑
1,8
‑
二醇。
[0056]
脂环族二醇的实例为环戊烷
‑
1,2
‑
和
‑
1,3
‑
二醇,环己烷
‑
1,2
‑
、
‑
1,3
‑
和
‑
1,4
‑
二醇,1,1
‑
、1,2
‑
、1,3
‑
和1,4
‑
双(羟甲基)环己烷,1,1
‑
、1,2
‑
、1,3
‑
和1,4
‑
双(羟乙基)环己烷和双(4
‑
羟基环己烷)亚异丙基。
[0057]
优选的脂环族二醇为环己烷
‑
1,2
‑
、
‑
1,3
‑
和
‑
1,4
‑
二醇,1,3
‑
和1,4
‑
双(羟甲基)环己烷和双(4
‑
羟基环己烷)亚异丙基。
[0058]
聚合二醇基本上是上述通式ho[ao]
m
h的聚氧化烯,其中ao为氧化乙烯、氧化丙烯、1,2
‑
环氧丁烷和/或2,3
‑
环氧丁烷,m为≥8的数。如果使用该聚合二醇来制备聚酯醇,则特别地使用相应的聚乙二醇。
[0059]
然而,为了制备聚酯醇,优选使用无环二醇乙二醇、丙烷
‑
1,2
‑
二醇、丙烷
‑
1,3
‑
二醇、丁烷
‑
1,4
‑
二醇、戊烷
‑
1,5
‑
二醇、己烷
‑
1,6
‑
二醇、2,2
‑
二甲基丙烷
‑
1,3
‑
二醇和/或辛烷
‑
1,8
‑
二醇。
[0060]
在二羧酸单元的情况下,有利的是使用游离二羧酸或其酸酐,这些可以具有芳族或脂族结构。在脂族二羧酸的情况下,使用具有无环结构的那些和具有脂环族结构的那些。
[0061]
具有无环结构的二羧酸的实例为c2‑
c
16
二酸,例如特别是草酸、丙二酸、马来酸、富马酸、琥珀酸、戊二酸、己二酸、庚二酸、辛二酸、壬二酸、癸二酸、十一烷二酸、十二烷二酸。
[0062]
脂环族二羧酸的实例为顺式
‑
和反式
‑
环己烷
‑
1,2
‑
二甲酸(六氢邻苯二甲酸),顺
式
‑
和反式
‑
环己烷
‑
1,3
‑
二甲酸,顺式
‑
和反式
‑
环己烷
‑
1,4
‑
二甲酸,1,2
‑
、1,3
‑
或1,4
‑
环己
‑4‑
烯二甲酸(四氢邻苯二甲酸),顺式
‑
和反式
‑
环戊烷
‑
1,2
‑
二甲酸,顺式
‑
和反式
‑
环戊烷
‑
1,3
‑
二甲酸。
[0063]
芳族二羧酸的实例为邻苯二甲酸、间苯二甲酸、对苯二甲酸和邻苯二甲酸酐,优选邻苯二甲酸和间苯二甲酸,特别优选邻苯二甲酸。
[0064]
有利地使用的二羧酸单元为琥珀酸、己二酸、癸二酸、马来酸、富马酸、邻苯二甲酸、间苯二甲酸、对苯二甲酸和/或邻苯二甲酸酐。
[0065]
根据本发明,特别有利的是基于己烷
‑
1,6
‑
二醇、乙二醇、亚丙基
‑
1,2
‑
二醇和/或新戊二醇作为二醇组分和己二酸、邻苯二甲酸、间苯二甲酸和/或癸二酸作为二羧酸组分的聚酯醇,其中特别优选基于己烷
‑
1,6
‑
二醇、己二酸和间苯二甲酸、己烷
‑
1,6
‑
二醇、己二酸和邻苯二甲酸的聚酯醇和/或基于亚丙基
‑
1,2
‑
二醇、丁烷
‑
1,4
‑
二醇和己二酸的聚酯醇。
[0066]
重要的是,分子和聚合化合物a均有利地可以由相应的上述包含羟基的分子化合物以及上述包含羟基的聚合化合物以简单的方式通过与适量的丙烯酰氯或甲基丙烯酰氯根据以下反应方程式反应,并以简单的方式除去氯化氢而得到:
[0067]
θ[
‑
oh]
n
+nch2=c(h/ch3)
‑
c(=o)
‑
cl
→
θ[
‑
o
‑
c(=o)
‑
c(h/ch3)=ch2]
n
+nhcl
[0068]
其中:
[0069]
θ:为至少二价的分子或聚合物有机基团,且
[0070]
n:为≥2的数。
[0071]
相应的反应是本领域技术人员所熟知的(参见例如r.jantas,s.polowinski:esterfication of poly(vinyl alcohol)with methacryloyl chloride,acta polym.第35卷,第2期,1984,第150
‑
152页)。
[0072]
分子以及聚合化合物a可在反应条件下通过使上述分子或聚合含羟基化合物与适量的丙烯酰氯或甲基丙烯酰氯在无溶剂下或有利地在本领域技术人员熟知的惰性有机溶剂存在下反应而制备。所用的惰性有机溶剂特别是其中所用的分子或聚合含羟基化合物和丙烯酰氯或甲基丙烯酰氯以及所得分子或聚合化合物a的量在反应温度下至少部分,有利地完全可溶,并且在反应温度下不与含羟基的化合物、丙烯酰氯或甲基丙烯酰氯以及所得化合物a反应的那些有机溶剂。所用的该有机溶剂的实例是极性非质子有机溶剂,其在大气压力(1.013绝对巴)下具有≥40℃且≤170℃,有利地≥60℃且≤130℃的沸点,例如特别是酮,例如丙酮、甲基乙基酮、甲基异丁基酮、甲基叔丁基酮;链烷酸烷基酯,例如乙酸乙酯、乙酸正丙酯、乙酸异丙酯、乙酸正丁酯、乙酸叔丁酯、丙酸甲酯或丙酸乙酯。
[0073]
在另一实施方案中,分子和聚合化合物a原则上也可通过相应的分子或聚合二羧酸或多羧酸化合物与(甲基)丙烯酸缩水甘油酯根据以下反应方程式反应而获得:
[0074][0075]
其中θ和n具有上述定义。在这种情况下,反应可以在没有溶剂的情况下进行,有利地在合适的有机溶剂中进行。
[0076]
在有利的实施方案中,在制备分子或聚合化合物a之后,以本领域技术人员熟知的方式(加入水并蒸出有机溶剂)用水代替有机溶剂,其中形成所述至少一种化合物a的水溶
液、乳液和/或分散体。
[0077]
除了所述至少一种有机化合物a以外,本发明的水性粘合剂配制剂还包含羧酸酰肼b。
[0078]
根据本发明,所用的羧酸酰肼b为单羧酸酰肼,或有利地为二羧酸二酰肼,其衍生自脂族、芳族和杂环单和二羧酸。在这种情况下,单羧酸单酰肼和二羧酸二酰肼有利地通过使相应的单羰基卤化物和二羰基二卤化物,特别是相应的单羰基氯化物和二羰基二氯化物与肼反应而制备。
[0079]
根据本发明可以使用的单羧酸酰肼b有利地为下式的化合物:
[0080]
r3‑
c(=o)
‑
nh
‑
nh2[0081]
其中r3为氢或单价c1‑
c
18
烷基、c6‑
c
12
芳基、c5‑
c
12
环烷基或具有至少一个氧、氮和/或硫原子的5
‑
6元杂环基,其中这些基团还可具有官能团如c1‑
c
18
烷基、c6‑
c
12
芳基、c5‑
c
12
环烷基、卤素或具有至少一个氧、氮和/或硫原子的5
‑
6元杂环基。
[0082]
脂族单羧酸酰肼b的实例为脂环族化合物甲酰肼[hc(=o)
‑
nh
‑
hn2]、乙酰肼[h3c
‑
c(=o)
‑
nh
‑
hn2]、丙酰肼[h3c
‑
ch2‑
c(=o)
‑
nh
‑
hn2]、丁酰肼[h3c
‑
(ch2)2‑
c(=o)
‑
nh
‑
hn2]、辛酰肼[h3c
‑
(ch2)3‑
c(=o)
‑
nh
‑
hn2],脂环族化合物环戊基甲酸酰肼或环己基甲酸酰肼,以及酰胺草酸酰肼[h2n
‑
c(=o)
‑
(c=o)
‑
nh
‑
nh2],芳族化合物苯甲酸酰肼、2
‑
氯苯甲酸酰肼、2
‑
硝基苯甲酸酰肼、3
‑
溴苯甲酸酰肼、4
‑
氯苯甲酸酰肼、4
‑
硝基苯甲酸酰肼,4
‑
溴苯甲酸酰肼,4
‑
氨基苯甲酸酰肼、4
‑
羟基苯甲酸酰肼、4
‑
甲基苯甲酸酰肼、苯乙酸酰肼、水杨酸酰肼、l
‑
酪氨酸酰肼、3
‑
羟基萘甲酸酰肼、烟酸酰肼、吡咯烷
‑2‑
甲酸酰肼或1
‑
乙酰基
‑2‑
苯基肼。
[0083]
优选的单羧酸酰肼b为乙酰肼和/或苯甲酸酰肼,特别优选乙酰肼。
[0084]
根据本发明可以使用的二羧酸二酰肼b有利地为下式的化合物:
[0085]
h2n
‑
nh
‑
c(=o)
‑
r4‑
c(=o)
‑
nh
‑
nh2[0086]
其中r4为具有1
‑
20个碳原子的无环、脂环族、芳族或杂环二价基团。
[0087]
r4例如为亚甲基、1,2
‑
亚乙基、1,2
‑
亚丙基、1,3
‑
亚丙基、1,2
‑
亚丁基、1,4
‑
亚丁基、1,3
‑
亚丁基、1,6
‑
亚己基、1,8
‑
亚辛基、1,12
‑
亚十二烷基、1,2
‑
亚苯基、1,3
‑
亚苯基、1,4
‑
亚苯基、1,2
‑
亚萘基、1,3
‑
亚萘基、1,4
‑
亚萘基、1,6
‑
亚萘基、1,2
‑
亚环戊基、1,3
‑
亚环戊基、1,2
‑
亚环己基、1,3
‑
亚环己基或1,4
‑
亚环己基。
[0088]
脂族二羧酸二酰肼b的实例为无环化合物草酸二酰肼,丙二酸二酰肼,琥珀酸二酰肼,戊二酸二酰肼和己二酸二酰肼。芳族二羧酸二酰肼的实例为邻苯二甲酸二酰肼、间苯二甲酸二酰肼或对苯二甲酸二酰肼。
[0089]
重要的是,根据本发明,碳酸的二酰肼—碳酸二酰肼[h2n
‑
nh
‑
c(=o)
‑
nh
‑
nh2]也可用作二酰肼化合物。
[0090]
优选的二羧酸二酰肼b为脂族二羧酸的二酰肼,特别优选丙二酸二酰肼或己二酸二酰肼。
[0091]
根据本发明,有利地使用在20℃和1.013巴(绝对)下溶解度≥5g/l,优选≥10,特别优选≥100g/l去离子水的羧酸酰肼b。任选地,为了提高羧酸酰肼b的溶解度,脂族醇,例如特别是甲醇、乙醇和/或异丙醇,可另外以≤5重量%,优选≤3重量%的量使用,在每种情况下基于水性粘合剂配制剂。
[0092]
在本发明的水性粘合剂配制剂中,选择有机化合物a和羧酸酰肼b的类型和量,以
使得所述至少一种化合物a的任选取代的丙烯酰氧基与所述至少一种羧酸酰肼b的酰肼基的当量摩尔比为≥0.01且≤10,有利地为≥0.5且≤5,优选为≥0.8且≤3.3,特别优选为≥1且≤3。
[0093]
重要的是,本发明的水性粘合剂配制剂包含≥20重量%,有利地≥30重量%,特别有利地≥30重量%的水和≤5重量%,有利地≤3重量%,特别有利地≤1重量%的有机溶剂。相应地,本发明的粘合剂配制剂的由所述至少一种有机化合物a与所述至少一种羧酸酰肼b的总和形成的活性粘合剂组分的含量为≥5重量%且≤75重量%,有利地≥10重量%且≤70重量%,特别有利地≥30重量%且≤60重量%。
[0094]
本发明的水性粘合剂配制剂是储存稳定的,并且在其施加时能够通过在干燥期间除去水而硬化,这是它们可以有利地用作粘合剂以制备胶粘剂、密封剂、打底涂料、纸张涂料浆、纤维非织造织物、柔性屋顶涂料、印刷油墨和涂料组合物中,以及用于砂子固结中,用作纺织品或皮革助剂和冲击改性剂的制备中的组分或者用于改性无机粘合剂和塑料的原因。
[0095]
所述水性粘合剂配制剂可以以简单的方式通过在水性介质中以任何顺序均匀混合有机化合物a和羧酸酰肼b来制备,其中制备可以预先进行或者在水性粘合剂配制剂的使用期间进行。有利地在其使用之前制备水性粘合剂配制剂。
[0096]
在特别优选的实施方案中,以如下方式制备水性粘合剂配制剂:在第一步中通过用水代替有机溶剂来制备所述至少一种化合物a的水溶液、乳液和/或分散体,并且在随后的第二步中将所述至少一种羧酸酰肼b加入到所述至少一种化合物a的该水溶液、乳液和/或分散体中。第二步有利地在均匀混合下进行。
[0097]
当然,在本发明的上下文中,取决于预期用途,所述水性粘合剂配制剂还可以包含其他常规助剂,其类型和量是本领域技术人员所熟知的,例如颜料、填料、染料、荧光增白剂、助留剂、润湿剂、成膜助剂、消泡剂、防腐剂、生物杀伤剂、粘液调节剂、增塑剂、防粘连剂、抗静电剂、缓冲物质、疏水剂等。
[0098]
本发明的水性粘合剂配制剂特别适合作为用于纤维和颗粒状基材的粘合剂。
[0099]
纤维和颗粒状基材是本领域技术人员所熟知的。例如,这些为木屑、木纤维、纺织纤维、玻璃纤维、矿物纤维或天然纤维,例如黄麻、亚麻、大麻或剑麻,但也可为软木屑或沙子。不言而喻,术语基材还应包括可由所述纤维获得的纤维非织造物,例如所谓的针刺纤维非织造物。本发明的水性粘合剂配制剂特别有利地适合作为用于前述天然纤维或由其形成的纤维非织造物的无甲醛粘合剂体系。
[0100]
使用水性粘合剂配制剂由纤维或颗粒状基材制备成型体的方法以如下方式进行:首先使纤维状或颗粒状基材与水性粘合剂配制剂接触(浸渍),然后使浸渍的纤维或颗粒状基材形成所需的形状,然后干燥或固化该形式。
[0101]
纤维和颗粒状基材通常以如下方式浸渍:将本发明的水性粘合剂配制剂均匀地施加到纤维和颗粒状基材的表面。在这种情况下,选择水性粘合剂配制剂的量,以使得每100g基材,可使用≥1g且≤100g,优选≥5g且≤50g,特别优选≥10g且≤30g的水性粘合剂配制剂,以固体形式计算(由有机化合物a和羧酸酰肼b的总和形成)。纤维和颗粒状基材的浸渍是本领域技术人员所熟知的,并且例如通过浸渍或通过喷涂纤维或颗粒状基材来进行。
[0102]
在浸渍后,例如通过将纤维或颗粒状基材置于可加热的压机或模具中,然后以本
领域技术人员熟知的方式干燥或固化,而使其成为所需的形状。
[0103]
所得成型体通常在≥0℃且≤130℃,优选≥10℃且≤100℃,特别优选≥15℃且≤50℃的温度下干燥和固化。
[0104]
在这种情况下,干燥和固化有利地以如下方式进行,即将成型体在规定的温度下干燥,直至得到的成型体具有≤10重量%,优选≤3重量%,特别优选≤0.5重量%的残余水分,其中粘合剂配制剂由于化学反应而固化。此处,残余水分通过首先在室温下称量得到的成型体,然后将其在130℃下干燥2分钟,然后将其冷却并在室温下再次称量来测定。此处,残余水分对应于干燥工艺前后成型体的重量差乘以因子100,基于干燥工艺前的成型体的重量。
[0105]
通过本发明方法得到的成型体具有有利的性能,特别是与现有技术的成型体相比,具有改善的弯曲变形和弯曲应力性能。
[0106]
因此,本发明特别地包括以下实施方案:
[0107]
1.一种水性粘合剂配制剂,其包含:
[0108]
a)至少一种有机化合物a,和
[0109]
b)至少一种羧酸酰肼b,
[0110]
其中所述至少一种有机化合物a
[0111]
a1)具有至少两个任选取代的丙烯酰氧基,且
[0112]
a2)不包含任何醛和/或酮羰基。
[0113]
2.根据实施方案1的水性粘合剂配制剂,其中所述任选取代的丙烯酰氧基具有以下结构:
[0114]
‑
o
‑
c(=o)
‑
chr1=chr2[0115]
其中:
[0116]
r1为氢、c1‑
c4烷基、
‑
c(=o)
‑
o
‑
c1‑
c4烷基或
‑
ch2‑
c(=o)
‑
o
‑
c1‑
c4烷基,且
[0117]
r2为氢、c1‑
c4烷基、
‑
c(=o)
‑
o
‑
c1‑
c4烷基或
‑
c≡n。
[0118]
3.根据实施方案1或2的水性粘合剂配料,其中所述至少一种化合物a为聚合化合物。
[0119]
4.根据实施方案1
‑
3中任一项的水性粘合剂配制剂,其中所用的至少一种羧酸酰肼b为脂族二羧酸的二酰肼。
[0120]
5.根据实施例1
‑
4中任一项的水性粘合剂配制剂,其中选择所述至少一种化合物a和所述至少一种羧酸酰肼b的量,以使得所述至少一种化合物a的任选取代的丙烯酰氧基与所述至少一种羧酸酰肼b的酰肼基的当量摩尔比为≥0.8且≤3.3。
[0121]
6.根据实施方案1
‑
5中任一项的水性粘合剂配料,其中所述配制剂包含≥20重量%的水和≤5重量%的有机溶剂。
[0122]
7.一种制备根据实施方案1
‑
6中任一项的水性粘合剂配制剂的方法,其中在第一步中制备所述至少一种化合物a的水溶液、乳液和/或分散体,并在随后的第二步中将所述至少一种羧酸酰肼b加入到所述至少一种化合物a的该水溶液、乳液和/或分散体中。
[0123]
8.根据实施方案1
‑
6中任一项的水性粘合剂配制剂作为粘合剂在胶粘剂、密封剂、打底涂料、纸张涂料浆、纤维非织造织物、柔性屋顶涂料、印刷油墨和涂料组合物的制备中以及在沙子固结中,作为纺织品或皮革助剂和抗冲改性剂的制备中的组分或改性无机粘合
剂和塑料的用途。
[0124]
9.根据实施方案1
‑
6中任一项的水性粘合剂配制剂作为颗粒和/或纤维状基材的粘合剂的用途。
[0125]
参考以下非限制性实施例更详细地描述本发明。
[0126]
所用的原料:
[0127]
1000/1:聚丙二醇(摩尔质量:1970g/mol;basf se产品)
[0128]
7600/1:脂族二醇、己二酸和芳族二羧酸的聚酯多元醇;摩尔质量:2000g/mol;basf se产品
[0129]
7800/1:脂族二醇、己二酸和芳族二羧酸的聚酯多元醇;摩尔质量:1000g/mol;basf se产品
[0130]
lr8765:丁二醇二缩水甘油醚二丙烯酸酯(basf se产品)
[0131]
3700:双酚a二缩水甘油醚二丙烯酸酯(allenex sa产品)
[0132]
mehq:4
‑
甲氧基苯酚
[0133]
bht:稳定剂,2,6
‑
二叔丁基对甲酚(basf se产品)
[0134]
tempol:稳定剂,4
‑
羟基
‑
2,2,6,6
‑
四甲基哌啶氧基
[0135]
kat 315:基于新癸酸铋的催化剂(borchers gmbh产品)
[0136]
ipdi:异佛尔酮二异氰酸酯
[0137]
hi 100:六亚甲基二异氰酸酯的三聚异氰脲酸酯(basf se产品)
[0138]
pud盐:(2
‑
氨基乙基)
‑3‑
氨基丙酸钠盐
[0139]
pe 1330:增稠剂(约30重量%的聚醚水溶液;basf se产品)聚氨酯水分散体的制备
[0140]
分散体1(d1)
[0141]
在氮气下,在20
‑
25℃(室温)下,首先将245.5g lupranol 1000/1、15.9g丙烯酸羟乙酯、14.2g laromer lr 8765、14.2g ebecryl 3700、23.4g 1,4
‑
丁二醇、1.8g正戊醇、21.2g 2,2
‑
(二羟甲基)丙酸、37.4g甲基乙基酮、0.5g kerobit bht、0.05g tempol和0.33g borchi kat 315加入到2l玻璃反应器中,并在搅拌下加热。当达到53℃的内部温度时,经5分钟滴加161.0g异佛尔酮二异氰酸酯,用17.3g甲基乙基酮洗涤进料管线,将反应混合物在油浴温度为100℃的油浴中搅拌。在达到114℃的最高内部温度后3小时,再向反应混合物中加入0.3g borchi kat 315。然后,将反应混合物在上述油浴温度下再搅拌4小时。在移除油浴并且不进一步加热之后,将反应混合物在搅拌下用483.2g丙酮稀释。在取出0.5g样品后,测得反应混合物的nco含量为0.27重量%。然后,将以此方式获得的反应混合物转移至4l蒸馏装置中并加热至52℃的内部温度。在不进一步加热下,经5分钟加入15.8g二乙基乙醇胺,将混合物再搅拌5分钟。然后,在20
‑
25℃下,在搅拌下经15分钟添加748.4g去离子水。然后,在100毫巴(绝对)压力下在搅拌下蒸出丙酮,直至达到43℃的内部温度。最后,将获得的反应混合物冷却至室温。
[0142]
所得聚氨酯分散体的固含量为39.7重量%,ph为7.5。测得平均粒度为672nm。
[0143]
在该文献的上下文中,nco含量通过将反应混合物的样品称重到250ml玻璃烧杯中
并在搅拌下用约150ml丙酮溶解而测定。使用分配器计量加入10ml 0.1m二乙基丁胺于n
‑
甲基
‑2‑
吡咯烷酮(nmp)中的溶液。然后将其在室温下使用获自metrohm的848titrino plus设备在搅拌下用0.1m盐酸反滴定。
[0144]
在该文献的上下文中,固含量通常通过使用mettler toledo hr73湿度分析仪在130℃的温度下将确定量的聚氨酯水分散体(约0.8g)干燥至恒重来测定。在每种情况下进行两次测量,并报告这两次测量的平均值。
[0145]
平均粒度通常根据iso 13321使用malvern high performance particle sizer在22℃和633nm波长下测定。
[0146]
ph通常通过校准的获自mettler
‑
toledo gmbh的325x ph电极测定。
[0147]
分散体2(d2)
[0148]
d2的制备与d1的制备完全类似,不同之处在于首先加入265.9g lupraphen 7600/1,62.2g丙烯酸羟乙酯,25.3g 2,2
‑
(二羟甲基)丙酸,39.3g甲基乙基酮,0.5g kerobit bht,0.05g tempol和0.65g borchi kat 315并加热。在46℃的内部温度开始,经5分钟滴加137.6g异佛尔酮二异氰酸酯,然后用14.8g甲基乙基酮洗涤进料管线。在达到104℃的最高内部温度后3.5小时,用476.7g丙酮稀释反应混合物(不进一步加入催化剂且不进一步加热)。测得nco含量为0.24重量%。在将反应混合物转移至4l蒸馏装置后,将22.0g二乙基乙醇胺和755.1g去离子水加入到反应混合物中。
[0149]
蒸馏后获得的聚氨酯分散体的固含量为40.5重量%,ph为7.3。平均粒度测定为56nm。
[0150]
分散体3(d3)
[0151]
d3的制备与d1的制备完全类似,不同之处在于首先加入288.8g lupraphen 7600/1、65.7g laromer lr 8765,3.9g正戊醇,19.8g 2,2
‑
(二羟甲基)丙酸、40.0g甲基乙基酮、0.5g kerobit bht、0.05g tempol和0.65g borchi kat 315并加热。在41℃的内部温度开始,经5分钟滴加117.6g异佛尔酮二异氰酸酯,然后用12.5g甲基乙基酮洗涤进料管线。在达到100℃的最高内部温度后4.5小时,将481.4g丙酮加入到反应混合物中(不进一步加入催化剂且不进一步加热)。测得nco含量为0.31重量%。在将反应混合物转移至4l蒸馏装置后,将17.2g二乙基乙醇胺和749.9g去离子水加入到反应混合物中。
[0152]
蒸馏后获得的聚氨酯分散体的固含量为36.1重量%,ph为7.9。平均粒度测定为41nm。
[0153]
分散体4(d4)
[0154]
d4的制备与d1的制备完全类似,不同之处在于首先将192.0g lupraphen 7600/1、53.4g ebecryl 3700、42.8g 1,4
‑
丁二醇、3.3g 1
‑
戊醇、16.2g(二羟甲基)丙酸、34.2g甲基乙基酮、0.5g kerobit bht、0.05g tempo和0.7g borchi kat 315加入到2l反应器中并加热。在61℃的内部温度开始,经5分钟滴加191.5g异佛尔酮二异氰酸酯,然后用20.7g甲基乙基酮洗涤进料管线。在达到126℃的最高内部温度后3小时15分钟,用484.5g丙酮稀释反应混合物(不进一步加入催化剂且不进一步加热)。测得nco含量为0.37重量%。在将反应混合物转移至4l蒸馏装置后,将13.9g二乙基乙醇胺和746.4g去离子水加入到反应混合物中。
[0155]
蒸馏后获得的聚氨酯分散体的固含量为39.8重量%,ph为7.5。平均粒度测定为36nm。
[0156]
分散体5(d5)
[0157]
d5的制备与d1的制备完全类似,不同之处在于首先将300.6g lupraphen 7600/1、4.2g丙烯酸羟乙酯、63.0g ebecryl 3700、22.6g(二羟甲基)丙酸、43.4g甲基乙基酮、0.50g kerobit bht、0.05g tempo和0.67g borchi kat 315加入到2l反应器中并加热。在50℃的内部温度开始,经5分钟滴加109.0g异佛尔酮二异氰酸酯,然后用11.6g甲基乙基酮洗涤进料管线。在达到100℃的最大温度后3小时,再将0.3g borchi kat 315加入到反应混合物中。然后,再将反应混合物在上述油浴温度下搅拌2小时20分钟。在移除油浴后且不进一步加热,将反应混合物在搅拌下用484.7g丙酮稀释。测得nco含量为0.18重量%。在将反应混合物转移至4l蒸馏装置后,将13.7g二乙基乙醇胺和746.1g去离子水加入到反应混合物中。
[0158]
用另外200g去离子水稀释蒸馏后获得的聚氨酯分散体,然后其固含量为30.1重量%,ph为7.3。平均粒度测定为41nm。
[0159]
分散体6(d6)
[0160]
d6的制备与d1的制备完全类似,不同之处在于首先将255.0glupraphen 7600/1、3.4g丙烯酸羟乙酯、44.3g 1,4
‑
丁二醇,18.6g(二羟甲基)丙酸、35.7g甲基乙基酮、0.50g kerobit bht、0.05g tempo和0.67g borchi kat 315加入到2l反应器中并加热。在55℃的内部温度开始,经5分钟滴加180.4g异佛尔酮二异氰酸酯,然后用19.5g甲基乙基酮洗涤进料管线。在达到114℃的最高内部温度后2小时40分钟,用487.1g丙酮稀释反应混合物(不进一步加入催化剂且不进一步加热)。测得nco含量为0.28重量%。在将反应混合物转移至4l蒸馏装置后,将11.3g二乙基乙醇胺和743.5g去离子水加入到反应混合物中。
[0161]
蒸馏后获得的聚氨酯分散体的固含量为37.9重量%,ph为7.6。平均粒度测定为55nm。
[0162]
分散体7(d7)
[0163]
d7的制备与d1的制备完全类似,不同之处在于首先将179.4g lupraphen 7600/1、3.2g丙烯酸羟乙酯、48.7g laromer lr 8765、41.5g 1,4
‑
丁二醇、21.0g(二羟甲基)丙酸、32.6g甲基乙基酮、0.49g kerobit bht、0.05g tempol和0.66g borchi kat 315加入到2l反应器中并加热。在45℃的内部温度开始,经5分钟滴加201.0g异佛尔酮二异氰酸酯,然后用21.8g甲基乙基酮洗涤进料管线。在达到110℃的最高内部温度后3.5小时,用480.3g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.42重量%。在将反应混合物转移至4l蒸馏装置后,将18.3g二乙基乙醇胺和751.0g去离子水加入到反应混合物中。
[0164]
蒸馏后获得的聚氨酯分散体的固含量为40.4重量%,ph为8.1。平均粒度测定为36nm。
[0165]
分散体8(d8)
[0166]
d8的制备与d1的制备完全类似,不同之处在于首先将234.0g lupranol 1000/1、26.8g丙烯酸羟乙酯、33.5g 1,4
‑
丁二醇、21.5g(二羟甲基)丙酸、35.1g甲基乙基酮、0.49g kerobit bht、0.05g tempo和0.66g borchi kat 315加入到2l反应器中并加热。在60℃的内部温度开始,经5分钟滴加178.5g异佛尔酮二异氰酸酯,然后用19.3g甲基乙基酮洗涤进料管线。在达到110℃的最高内部温度后4小时,用479.9g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.29重量%。在将反应混合物转移至4l蒸馏装
置后,将18.7g二乙基乙醇胺和751.5g去离子水加入到反应混合物中。
[0167]
蒸馏后获得的聚氨酯分散体的固含量为37.3重量%,ph为7.9。平均粒度测定为424nm。
[0168]
分散体9(d9)
[0169]
d9的制备与d1的制备完全类似,不同之处在于首先将245.3g lupraphen 7600/1、14.6g丙烯酸羟乙酯、14.2g laromer lr 8765、14.2g ebecryl 3700、23.3g 1,4
‑
丁二醇、23.4g(二羟甲基)丙酸、37.2g甲基乙基酮、0.50g kerobit bht、0.05g tempol和0.66g borchi kat 315加入到2l反应器中并加热。在49℃的内部温度开始,经5分钟滴加160.8g异佛尔酮二异氰酸酯,然后用17.3g甲基乙基酮洗涤进料管线。在达到110℃的最高内部温度后5小时,用481.2g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.29重量%。在将反应混合物转移至4l蒸馏装置后,将17.4g二乙基乙醇胺和750.1g去离子水加入到反应混合物中。
[0170]
蒸馏后获得的聚氨酯分散体的固含量为38.8重量%,ph为7.6。平均粒度测定为66nm。
[0171]
分散体10(d10)
[0172]
d10的制备与d1的制备完全类似,不同之处在于首先将382.6g lupranol 1000/1、4.3g丙烯酸羟乙酯、21.4g(二羟甲基)丙酸、45.4g甲基乙基酮、0.49g kerobit bht、0.05g tempo和0.66g borchi kat 315加入到2l反应器中并加热。在46℃的内部温度开始,经5分钟滴加86.2g异佛尔酮二异氰酸酯,然后用9.0g甲基乙基酮洗涤进料管线。在达到100℃的最高内部温度后6小时15分钟,用480.1g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.16重量%。在将反应混合物转移至4l蒸馏装置后,将18.5g二乙基乙醇胺和851.3g去离子水加入到反应混合物中。
[0173]
将另外400g去离子水加入到蒸馏后获得的聚氨酯分散体中。所述分散体的固含量为29.3重量%,ph为8.0。平均粒度测定为26nm。
[0174]
分散体11(d11)
[0175]
d11的制备与d1的制备完全类似,不同之处在于首先将188.3g lupranol 1000/1、52.1g laromer lr 8765、41.7g 1,4
‑
丁二醇、3.0g 1
‑
戊醇、17.5g(二羟甲基)丙酸、33.6g甲基乙基酮、0.50g kerobit bht、0.05g tempol和0.67g borchi kat 315加入到2l反应器中并加热。在40℃的内部温度开始,经5分钟滴加199.7g异佛尔酮二异氰酸酯,然后用21.6g甲基乙基酮洗涤进料管线。在达到115℃的最高内部温度后3.5小时,用487.7g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.57重量%。在将反应混合物转移至4l蒸馏装置后,将10.7g二乙基乙醇胺和742.8g去离子水加入到反应混合物中。
[0176]
蒸馏后获得的聚氨酯分散体的固含量为41.7重量%,ph为7.7。平均粒度测定为684nm。
[0177]
分散体12(d12)
[0178]
d12的制备与d1的制备完全类似,不同之处在于首先将180.7g lupranol 1000/1、3.0g丙烯酸羟乙酯、49.1g ebecryl 3700、41.8g 1,4
‑
丁二醇、24.6g(二羟甲基)丙酸、33.2g甲基乙基酮、0.50g kerobit bht、0.05g tempol和0.66g borchi kat 315加入到2l反应器中并加热。在56℃的内部温度开始,经8分钟滴加198.9g异佛尔酮二异氰酸酯,然后
用21.6g甲基乙基酮洗涤进料管线。在达到104℃的最高内部温度后6.5小时,用483.5g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.34重量%。在将反应混合物转移至4l蒸馏装置后,将15.0g二乙基乙醇胺和747.5g去离子水加入到反应混合物中。
[0179]
蒸馏后获得的聚氨酯分散体的固含量为38.8重量%,ph为7.4。平均粒度测定为199nm。
[0180]
分散体13(d13)
[0181]
d13的制备与d1的制备完全类似,不同之处在于首先将277.2g lupranol 1000/1、65.9g ebecryl 3700、3.9g 1
‑
戊醇、26.8g(二羟甲基)丙酸、41.5g甲基乙基酮、0.49g kerobit bht、0.05g tempo和0.65g borchi kat 315加入到2l反应器中并加热。在47℃的内部温度开始,经5分钟滴加116.0g异佛尔酮二异氰酸酯,然后用12.3g甲基乙基酮洗涤进料管线。在达到85℃的最高内部温度后6小时,用475.5g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.22重量%。在将反应混合物转移至4l蒸馏装置后,将23.2g二乙基乙醇胺和756.5g去离子水加入到反应混合物中。此外,在蒸馏期间加入另外700g去离子水。
[0182]
蒸馏后获得的聚氨酯分散体的固含量为25.7重量%,ph为8.0。平均粒度测定为31nm。
[0183]
分散体14(d14)
[0184]
d14的制备与d1的制备完全类似,不同之处在于首先将276.0g lupraphen 7600/1、32.7g丙烯酸羟乙酯、12.4g 1,4
‑
丁二醇、26.3g(二羟甲基)丙酸、38.6g甲基乙基酮、0.49g kerobit bht、0.05g tempol和0.65g borchi kat 315加入到2l反应器中并加热。在52℃的内部温度开始,经5分钟滴加142.8g异佛尔酮二异氰酸酯,然后用15.3g甲基乙基酮洗涤进料管线。在达到105℃的最高内部温度后3小时15分钟,用475.9g丙酮稀释反应混合物(不进一步加入催化剂且不进一步加热)。测得nco含量为0.20重量%。在将反应混合物转移至4l蒸馏装置后,将22.8g二乙基乙醇胺和756.0g去离子水加入反应混合物中。
[0185]
蒸馏后获得的聚氨酯分散体的固含量为42.0重量%,ph为7.6。平均粒度测定为31nm。
[0186]
分散体15(d15)
[0187]
d15的制备与d1的制备完全类似,不同之处在于首先将213.9g lupraphen 7600/1、28.4g 1,4
‑
丁二醇、18.3g(二羟甲基)丙酸,47.2g laromer 8765、34.2g甲基乙基酮、0.49g kerobit bht和0.05g tempol加入到2l反应器中并加热。在48℃的内部温度开始,经5分钟滴加185.1g异佛尔酮二异氰酸酯,然后用20.0g甲基乙基酮洗涤进料管线。在达到95℃的最高内部温度后3小时,用471.3g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为1.10重量%。在将反应混合物转移至4l蒸馏装置后,将2.1g异佛尔酮二胺、16.0g二乙基乙醇胺、724.9g去离子水,以及5.7g二亚乙基三胺与32.4g去离子水的溶液依次加入到反应混合物中。
[0188]
蒸馏后获得的聚氨酯分散体的固含量为40.8重量%,ph为8.0。平均粒度测定为61nm。
[0189]
分散体16(d16)
[0190]
d16的制备与d1的制备完全类似,不同之处在于首先将204.1g lupraphen 7600/1、25.5g 1,4
‑
丁二醇、18.1g(二羟甲基)丙酸、69.3g ebecryl 3700、35.2g甲基乙基酮、0.49g kerobit bht和0.05g tempol加入到2l反应器中并加热。在50℃的内部温度开始,经5分钟滴加176.6g异佛尔酮二异氰酸酯,然后用19.1g甲基乙基酮洗涤进料管线。在达到101℃的最高内部温度后3小时,用472.0g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为1.17重量%。在将反应混合物转移至4l蒸馏装置后,将2.0g异佛尔酮二胺、15.9g二乙基乙醇胺、725.3g去离子水,以及5.5g二亚乙基三胺与31.0g去离子水的溶液加入到反应混合物中。
[0191]
蒸馏后获得的聚氨酯分散体的固含量为40.9重量%,ph为7.7。平均粒度测定为61nm。
[0192]
分散体17(d17)
[0193]
d17的制备与d1的制备完全类似,不同之处在于首先将277.1g lupraphen 7600/1、32.8g丙烯酸羟乙酯、16.3g 1,4
‑
丁二醇、20.6g(二羟甲基)丙酸、38.5g甲基乙基酮、0.49g kerobit bht、0.05g tempo和0.65g borchi kat 315加入到2l反应器中并加热。在49℃的内部温度开始,经5分钟滴加143.4g异佛尔酮二异氰酸酯,然后用15.4g甲基乙基酮洗涤进料管线。在达到102℃的最高内部温度后3小时,用475.8g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.23重量%。在将反应混合物转移至4l蒸馏装置后,将18.0g二乙基乙醇胺和756.1g去离子水加入到反应混合物中。
[0194]
蒸馏后获得的聚氨酯分散体的固含量为39.9重量%,ph为7.4。平均粒度测定为36nm。
[0195]
分散体18(d18)
[0196]
d18的制备与d1的制备完全类似,不同之处在于首先将250.8g lupraphen 7600/1、14.9g丙烯酸羟乙酯、14.5g laromer lr 8765、14.5g ebecryl 3700、23.9g 1,4
‑
丁二醇、23.9g(二羟甲基)丙酸、38.1g甲基乙基酮、0.51g kerobit bht、0.05g tempol和0.68g borchi kat 315加入到2l反应器中并加热。在49℃的内部温度开始,经5分钟滴加164.5g异佛尔酮二异氰酸酯,然后用17.7g甲基乙基酮洗涤进料管线。在达到105℃的最高内部温度后4小时45分钟,用492.2g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.25重量%。在将反应混合物转移至4l蒸馏装置后,将60.6g 10重量%氢氧化钠水溶液和683.3g去离子水加入到反应混合物中。此外,在蒸馏期间加入另外300g去离子水。
[0197]
蒸馏后获得的聚氨酯分散体的固含量为32.6重量%,ph为7.7。平均粒度测定为31nm。
[0198]
分散体19(d19)
[0199]
d19的制备与d1的制备完全类似,不同之处在于首先将283.5g lupraphen 7600/1、33.6g丙烯酸羟乙酯,20.5g 1,4
‑
丁二醇、15.4g(二羟甲基)丙酸、39.2g甲基乙基酮、0.50g kerobit bht、0.05g tempo和0.67g borchi kat 315加入到2l反应器中并加热。在52℃的内部温度开始,经5分钟滴加146.7g异佛尔酮二异氰酸酯,然后用15.8g甲基乙基酮洗涤进料管线。在达到100℃的最高内部温度后2.5小时,用485.1g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.23重量%。在将反应混合物转移至
4l蒸馏装置后,将13.4g二乙基乙醇胺和745.7g去离子水加入到反应混合物中。
[0200]
蒸馏后获得的聚氨酯分散体的固含量为42.4重量%,ph为7.7。平均粒度测定为449nm。
[0201]
分散体20(d20)
[0202]
d20的制备与d1的制备完全类似,不同之处在于首先将251.9g lupraphen 7600/1、15.0g丙烯酸羟乙酯、14.6g laromer lr 8765、14.6g ebecryl 3700、26.4g 1,4
‑
丁二醇、20.4g(二羟甲基)丙酸、38.1g甲基乙基酮、0.51g kerobit bht、0.05g tempol和0.68g borchi kat 315加入到2l反应器中并加热。在55℃的内部温度开始,经5分钟滴加165.1g异佛尔酮二异氰酸酯,然后用17.8g甲基乙基酮洗涤进料管线。在达到111℃的最高内部温度后4小时10分钟,用493.0g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.25重量%。在将反应混合物转移至4l蒸馏装置后,将51.7g 10重量%氢氧化钠水溶液和690.3g去离子水加入到反应混合物中。此外,在蒸馏期间加入另外350g去离子水。
[0203]
蒸馏后获得的聚氨酯分散体的固含量为32.6重量%,ph为7.9。平均粒度测定为35nm。
[0204]
分散体21(d21)
[0205]
d21的制备与d1的制备完全类似,不同之处在于首先将281.7g lupraphen 7600/1、33.4g丙烯酸羟乙酯、18.6g 1,4
‑
丁二醇、18.0g(二羟甲基)丙酸、39.1g甲基乙基酮、0.50g kerobit bht、0.05g tempo和0.66g borchi kat 315加入到2l反应器中并加热。在50℃的内部温度开始,经5分钟滴加145.8g异佛尔酮二异氰酸酯,然后用15.7g甲基乙基酮洗涤进料管线。在达到100℃的最高内部温度2小时后,用482.9g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.23重量%。在将反应混合物转移至4l蒸馏装置后,将15.6g二乙基乙醇胺和748.1g去离子水加入到反应混合物中。
[0206]
蒸馏后获得的聚氨酯分散体的固含量为42.5重量%,ph为7.5。平均粒度测定为309nm。
[0207]
分散体22(d22)
[0208]
d22的制备与d1的制备完全类似,不同之处在于首先将252.6g lupraphen 7600/1、15.0g丙烯酸羟乙酯、14.6g laromer lr 8765、14.6g ebecryl 3700、28.1g丁二醇、18.0g(二羟甲基)丙酸、38.1g甲基乙基酮、0.51g kerobit bht、0.05g tempo和0.68g borchi kat 315加入到2l反应器中并加热。在65℃的内部温度开始,经5分钟滴加165.6g异佛尔酮二异氰酸酯,然后用17.8g甲基乙基酮洗涤进料管线。在达到114℃的最高内部温度后4小时,用493.6g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.27重量%。在将反应混合物转移至4l蒸馏装置后,将45.6g 10重量%氢氧化钠水溶液和695.2g去离子水加入到反应混合物中。此外,在蒸馏期间加入另外250g去离子水。
[0209]
蒸馏后获得的聚氨酯分散体的固含量为34.3重量%,ph为8.1。平均粒度测定为39nm。
[0210]
分散体23(d23)
[0211]
d23的制备与d1的制备完全类似,不同之处在于首先将173.2g lupraphen 7600/1、23.0g(二羟甲基)丙酸、147.4g ebecryl 3700、38.2g甲基乙基酮、0.49g kerobit bht和
0.05g tempol加入到2l反应器中并加热。在50℃的内部温度开始,经5分钟滴加149.7g异佛尔酮二异氰酸酯,然后用16.1g甲基乙基酮洗涤进料管线。在达到100℃的最高内部温度后4小时,用471.7g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为1.00重量%。在将反应混合物转移至4l蒸馏装置后,将1.7g异佛尔酮二胺、18.0g二乙基乙醇胺、729.5g去离子水,以及4.6g二亚乙基三胺和26.3g去离子水的溶液加入到反应混合物中。此外,在蒸馏期间加入另外100g去离子水。
[0212]
蒸馏后获得的聚氨酯分散体的固含量为36.3重量%,ph为7.7。平均粒度测定为55nm。
[0213]
分散体24(d24)
[0214]
d24的制备与d1的制备完全类似,不同之处在于首先将379.1g lupraphen 7600/1、22.5g丙烯酸羟乙酯、21.9g laromer lr 8765、21.9g ebecryl 3700、33.2g 1,4
‑
丁二醇、40.4g(二羟甲基)丙酸、57.7g甲基乙基酮,0.77g kerobit bht、0.08g tempol和1.02g borchi kat 315加入到2l反应器中并加热。在50℃的内部温度开始,经5分钟滴加248.5g异佛尔酮二异氰酸酯,然后用26.8g甲基乙基酮洗涤进料管线。在达到107℃的最高内部温度后3小时,用745.2g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.26重量%(所得产物以下也称为预聚物)。在将800g反应混合物部分转移至4l蒸馏装置后,将15.4g三乙胺和589.3g去离子水加入到反应混合物中。此外,在蒸馏期间加入另外100g去离子水,在蒸馏之后加入另外200g去离子水。
[0215]
在这种情况下获得的聚氨酯分散体具有30.2重量%的固含量和7.3的ph。平均粒度测定为38nm。
[0216]
分散体25(d25)
[0217]
将730g来自实验24的预聚物称入4l蒸馏装置中并加热至53℃。将55.4g 10重量%氢氧化钠水溶液和487.8g去离子水加入反应混合物中,然后蒸馏该混合物。此外,在蒸馏期间加入另外200g去离子水。
[0218]
蒸馏后获得的聚氨酯分散体的固含量为35.1重量%,ph为7.5。平均粒度测定为38nm。
[0219]
分散体26(d26)
[0220]
d26的制备与d1的制备完全类似,不同之处在于首先将179.8g lupraphen 7600/1、5.0g丙烯酸羟乙酯、46.1g laromer lr 8765、41.5g 1,4
‑
丁二醇、21.0g(二羟甲基)丙酸、32.6g甲基乙基酮、0.49g kerobit bht、0.05g tempol和0.66g borchi kat 315加入到2l反应器中并加热。在57℃的内部温度开始,经5分钟滴加201.3g异佛尔酮二异氰酸酯,然后用21.8g甲基乙基酮洗涤进料管线。在达到117℃的最高内部温度后4小时,用480.3g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.36重量%。在将反应混合物转移至4l蒸馏装置后,将18.3g二乙基乙醇胺和751.1g去离子水加入到反应混合物中。此外,在蒸馏后,加入另外250g去离子水。
[0221]
在这种情况下获得的聚氨酯分散体具有29.1重量%的固含量和7.6的ph。平均粒度测定为35nm。
[0222]
分散体27(d27)
[0223]
d27的制备与d1的制备完全类似,不同之处在于首先将246.6g lupraphen 7600/
1、14.6g丙烯酸羟乙酯、14.3g laromer lr 8765、14.2g ebecryl 3700、21.6g 1,4
‑
丁二醇、26.6g(二羟甲基)丙酸、37.5g甲基乙基酮、0.50g kerobit bht、0.05g tempol和0.67g borchi kat 315加入到2l反应器中并加热。在50℃的内部温度开始,经5分钟滴加161.7g异佛尔酮二异氰酸酯,然后用17.4g甲基乙基酮洗涤进料管线。在达到106℃的最高内部温度后3小时,用484.7g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.28重量%。在将反应混合物转移至4l蒸馏装置后,将13.8g二乙基乙醇胺和746.2g去离子水加入到反应混合物中。
[0224]
蒸馏后获得的聚氨酯分散体的固含量为43.4重量%,ph为6.9。平均粒度测定为344nm。
[0225]
分散体28(d28)
[0226]
d28的制备与d1的制备完全类似备,不同之处在于首先将253.8g lupraphen 7600/1、15.1g丙烯酸羟乙酯、14.7g laromer lr 8765、14.7g ebecryl 3700、31.7g 1,4
‑
丁二醇、12.9g(二羟甲基)丙酸、38.1g甲基乙基酮、0.51g kerobit bht、0.05g tempol和0.68g borchi kat 315加入到2l反应器中并加热。在63℃的内部温度开始,经5分钟滴加166.4g异佛尔酮二异氰酸酯,然后用17.9g甲基乙基酮洗涤进料管线。在达到112℃的最高内部温度后4小时10分钟,用494.3g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.25重量%。在将反应混合物转移至4l蒸馏装置后,将38.5g 10重量%氢氧化钠水溶液和700.8g去离子水加入到反应混合物中。此外,在蒸馏期间加入另外50g去离子水。
[0227]
蒸馏后获得的聚氨酯分散体的固含量为39.3重量%,ph为8.0。平均粒度测定为87nm。
[0228]
分散体29(d29)
[0229]
d29的制备与d1的制备完全类似,不同之处在于首先将254.3g lupraphen 7600/1、15.1g丙烯酸羟乙酯、14.7g laromer lr 8765、14.7g ebecryl 3700、32.9g 1,4
‑
丁二醇、11.3g(二羟甲基)丙酸、38.1g甲基乙基酮、0.51g kerobit bht、0.05g tempol和0.68g borchi kat 315加入到2l反应器中并加热。在52℃的内部温度开始,经5分钟滴加166.7g异佛尔酮二异氰酸酯,然后用18.0g甲基乙基酮洗涤进料管线。在达到109℃的最高内部温度后3.5小时,用494.8g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.26重量%。在将反应混合物转移至4l蒸馏装置后,将33.7g 10重量%氢氧化钠水溶液和704.6g去离子水加入到反应混合物中。
[0230]
蒸馏后获得的聚氨酯分散体的固含量为42.8重量%,ph为8.0。平均粒度测定为175nm。
[0231]
分散体30(d30)
[0232]
d30的制备与d1的制备完全类似,不同之处在于首先将214.8g lupraphen 7600/1、30.5g 1,4
‑
丁二醇、15.7g(二羟甲基)丙酸、47.6g laromer 8765、34.3g甲基乙基酮、0.50g kerobit bht和0.05g tempol加入到2l反应器中并加热。在50℃的内部温度开始,经5分钟滴加186.4g异佛尔酮二异氰酸酯,然后用20.2g甲基乙基酮洗涤进料管线。在达到112℃的最高内部温度后3小时,用473.4g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为1.20重量%。在将反应混合物转移至4l蒸馏装置后,将2.15g异佛
尔酮二胺、13.7g二乙基乙醇胺、722.4g去离子水,以及5.8g二亚乙基三胺与32.6g去离子水的溶液加入反应混合物中。
[0233]
蒸馏后获得的聚氨酯分散体的固含量为41.2重量%,ph为7.7。平均粒度测定为115nm。
[0234]
分散体31(d31)
[0235]
d31的制备与d1的制备完全类似,不同之处在于首先将236.1g lupraphen 7600/1、31.5g 1,4
‑
丁二醇、18.0g(二羟甲基)丙酸、80.3g ebecryl 3700、40.6g甲基乙基酮、0.57g kerobit bht和0.06g tempol加入到2l反应器中并加热。在52℃的内部温度开始,经5分钟滴加204.8g异佛尔酮二异氰酸酯,然后用22.1g甲基乙基酮洗涤进料管线。在达到90℃的升高的内部温度(至多95℃)后4小时40分钟,用545.8g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为1.17重量%。在将反应混合物转移至4l蒸馏装置后,将2.4g异佛尔酮二胺、15.8g二乙基乙醇胺、559.8g去离子水,以及6.3g二亚乙基三胺与35.8g去离子水的溶液加入到反应混合物中。
[0236]
蒸馏后获得的聚氨酯分散体的固含量为51.2重量%,ph为7.5。平均粒度测定为350nm。
[0237]
分散体32(d32)
[0238]
d32的制备与d1的制备完全类似,不同之处在于首先将187.9g lupraphen 7600/1、23.4g(二羟甲基)丙酸、118.1g laromer lr 8765、36.6g甲基乙基酮、0.49g kerobit bht和0.05g tempo加入到2l反应器中并加热。在45℃的内部温度开始,经5分钟滴加162.6g异佛尔酮二异氰酸酯,然后用17.5g甲基乙基酮洗涤进料管线。在达到102℃的最高内部温度后2小时10分钟,用470.5g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为1.06重量%。在将反应混合物转移至4l蒸馏装置后,将1.8g异佛尔酮二胺、18.5g二乙基乙醇胺、729.1g去离子水,以及5.0g二亚乙基三胺与28.5g去离子水的溶液加入到反应混合物中。此外,在蒸馏期间加入另外300g去离子水。
[0239]
蒸馏后获得的聚氨酯分散体的固含量为31.5重量%,ph为7.7。平均粒度测定为72nm。
[0240]
分散体33(d33)
[0241]
d33的制备与d1的制备完全类似,不同之处在于首先91.5g lupraphen 7800/1、119.1g甲基丙烯酸羟乙酯、44.1g 1,4
‑
丁二醇、200.1g丙酮、0.80g kerobit bht、0.05g tempol、0.47g 4
‑
甲氧基苯酚和0.57g borchi kat 315加入到2l反应器中并加热。在48℃的内部温度开始,经15分钟滴加215.8g异佛尔酮二异氰酸酯和64.8g basonat hi 100。在达到70℃的最高内部温度后2小时,测得nco含量为1.28重量%。在将反应混合物转移至4l蒸馏装置中并用49.9g丙酮洗涤后,在48℃的温度下将43.1g 40重量%的pud盐水溶液加入到反应混合物中并用669.8g去离子水分散。
[0242]
蒸馏后获得的聚氨酯分散体的固含量为43.8重量%,ph为8.0。平均粒度测定为82nm。
[0243]
分散体34(d34)
[0244]
d34的制备与d1的制备完全类似,不同之处在于首先将244.4g lupraphen 7600/1、16.3g甲基丙烯酸羟乙酯、14.1g laromer lr 8765、14.1g ebecryl 3700、23.3g 1,4
‑
丁
二醇、23.3g(二羟甲基)丙酸、37.3g甲基乙基酮、0.50g kerobit bht、0.05g tempol和0.66g borchi kat 315加入到2l反应器中并加热。在52℃的内部温度开始,经5分钟滴加160.2g异佛尔酮二异氰酸酯,然后用17.3g甲基乙基酮冲洗进料管线。在达到100℃的最高内部温度后5小时,用481.5g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.28重量%。在将反应混合物转移至4l蒸馏装置后,将17.3g二乙基乙醇胺和750.0g去离子水加入到反应混合物中。此外,在蒸馏后加入另外100.0g去离子水。
[0245]
在这种情况下获得的聚氨酯分散体具有39.0重量%的固含量和7.5的ph。平均粒度测定为84nm。
[0246]
分散体35(d35)
[0247]
d35的制备与d1的制备完全类似,不同之处在于首先将202.4g lupraphen 7600/1、23.9g(二羟甲基)丙酸、18.0g丙烯酸羟乙酯、38.7g甲基乙基酮、0.50g kerobit bht和0.05g tempo加入到2l反应器中并加热。在43℃的内部温度开始,经5分钟滴加154.9g异佛尔酮二异氰酸酯,然后用16.7g甲基乙基酮洗涤进料管线。在供入异佛尔酮二异氰酸酯后1小时25分钟,加入104.1g laromer lr 8765。在达到86℃的升高的温度后8.5小时,用481.2g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.39重量%。在将反应混合物转移至4l蒸馏装置后,将18.8g二乙基乙醇胺和740.9g去离子水加入到反应混合物中。另外,在蒸馏期间加入另外250g去离子水。
[0248]
蒸馏后获得的聚氨酯分散体的固含量为32.9重量%,ph为7.2。平均粒度测定为36nm。
[0249]
分散体36(d36)
[0250]
d36的制备与d1的制备完全类似,不同之处在于首先将279.4g lupraphen 7600/1、22.3g(二羟甲基)丙酸、72.8g ebecryl 3700、41.6g甲基乙基酮、0.50g kerobit bht和0.05g tempol加入到2l反应器中并加热。在60℃的内部温度开始,经5分钟滴加121.5g异佛尔酮二异氰酸酯,然后用13.0g甲基乙基酮洗涤进料管线。在达到102℃的最高内部温度后3小时15分钟,用474.2g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.85重量%。在将反应混合物转移至4l蒸馏装置后,将1.4g异佛尔酮二胺、17.5g二乙基乙醇胺、731.0g去离子水,以及3.8g二亚乙基三胺与21.3g去离子水的溶液加入到反应混合物中。此外,在蒸馏期间加入另外50g去离子水。
[0251]
蒸馏后获得的聚氨酯分散体的固含量为38.0重量%,ph为8.0。平均粒度测定为31nm。
[0252]
分散体37(d37)
[0253]
d37的制备与d1的制备完全类似,不同之处在于首先将273.9g lupraphen 7600/1、36.4g甲基丙烯酸羟乙酯,12.3g 1,4
‑
丁二醇,26.1g(二羟甲基)丙酸,38.7g甲基乙基酮,0.49g kerobit bht,0.05g tempo和0.65g borchi kat 315加入到2l反应器中并加热。在52℃的内部温度开始,经5分钟滴加141.8g异佛尔酮二异氰酸酯,然后用15.2g甲基乙基酮洗涤进料管线。在达到105℃的最高内部温度后2.5小时,用476.1g丙酮稀释反应混合物(不进一步添加催化剂且不进一步加热)。测得nco含量为0.25重量%。在将反应混合物转移至4l蒸馏装置后,将22.6g二乙基乙醇胺和755.8g去离子水加入到反应混合物中。
[0254]
蒸馏后获得的聚氨酯分散体的固含量为46.2重量%,ph为7.6。平均粒度测定为
45nm。
[0255]
对比实施例1(c1)
[0256]
c1的制备与d1的制备完全类似,不同之处在于首先将114.6g lupraphen 7800/1、68.9g 1
‑
丙醇、55.3g 1,4
‑
丁二醇、187.6g丙酮、0.89g kerobit bht、0.06g tempo、0.51g mehq和0.39g borchi kat 315加入到2l反应器中并加热。在49℃的内部温度开始,经15分钟滴加270.4g异佛尔酮二异氰酸酯和81.1g basonat hi 100。在加入异佛尔酮二异氰酸酯和basonate hi 100后1小时35分钟,再加入0.2g borchi kat 315。在68
‑
73℃的恒定内部温度下4小时24分钟的总反应时间后,测得nco含量为1.54重量%。在将反应混合物转移至4l蒸馏装置中并用88.4g丙酮洗涤后,在49℃下将54.1g 40重量%的pud盐水溶液加入到反应混合物中,并用另外877.5g去离子水分散。此外,在蒸馏期间加入另外400g去离子水。
[0257]
蒸馏后获得的聚氨酯分散体的固含量为34.0重量%,ph为9.5。平均粒度测定为126nm。
[0258]
性能测试
[0259]
测定膜硬度
[0260]
在室温和搅拌下,在每种情况下向聚氨酯分散体d1
‑
d37和c1的测试部分中加入2.5重量%己二酸二酰肼(addh),在每种情况下基于相应聚氨酯分散体的聚氨酯固含量。使用聚氨酯分散剂d1
‑
d37和c1的另一测试部分,但不加入addh。如果在这种情况下,含添加剂的聚氨酯分散体或不含添加剂的聚氨酯分散体的流动时间小于20秒(使用erichsen din烧杯4根据din 53211测定),则加入足够的rheovis pe 1330,直至其粘度为≥20且≤60秒(典型的施加水平)。
[0261]
在其制备后或使用前,立即使用speedmixer dac 150.1 fvz将所得的含有或不含添加剂的聚氨酯水分散体d1
‑
d37和c1在3500rpm下均匀混合5分钟。然后,在室温下用300μm盒型涂布棒将所得的含有或不含添加剂的聚氨酯水分散体施加至不含油脂的10
×
8cm玻璃板上。如下表1所述,将所得湿膜在室温和60%相对湿度下在气候调节室中在黑暗中干燥并储存同样如表1所述的时间,或者在所述温度下干燥并成膜30分钟,然后在气候调节室中在黑暗中在室温和60%相对湿度下储存同样如表中所述的时间。在这种情况下,干燥和储存后获得的膜层厚度为40
‑
100μm。
[0262]
根据din en iso 1522测定干燥和储存后获得的聚合物膜的摆阻尼k。在这种情况下,同样如表1所述的秒数越高,则评价所得聚合物膜越硬。
[0263]
表1:摆阻尼测量结果
[0264][0265]
[0266]
从以上结果清楚地看出,与由相应的不含添加剂的聚氨酯分散体制备的聚合物膜相比,由具有addh添加剂的丙烯酰氧基官能化聚氨酯制备的所有聚合物膜最晚在6周的储存时间之后具有提高的摆测硬度。
[0267]
对比分散体c1的聚氨酯不具有任何丙烯酰氧基。相应结果的显著之处在于,在这种情况下,不含添加剂的聚氨酯分散体的膜硬度甚至高于含有addh添加剂的聚氨酯分散体的相应膜硬度,这可例如由在聚合物膜中包含未反应的addh来解释。
[0268]
不同酰肼化合物的使用
[0269]
为此,在每种情况下,在室温下将0.5g rheovis pe1330和合适的酰肼化合物加入到50g聚氨酯分散体d33中。随后立即使用speedmixer dac 150.1 fvz将各混合物在3500rpm下均匀混合5分钟。所用的酰肼化合物为0.51g己二酸二酰肼(d33
‑
1)、0.29g碳酸二酰肼(d33
‑
2)、0.57g间苯二甲酸二酰肼(d33
‑
3)和0.44g乙酰肼(d33
‑
4)。50g聚氨酯分散体d33与0.5g rheovis pe1330而不加入酰肼的混合物用作对比(d33
‑
c)。
[0270]
在其制备后,立即使用300μm盒型涂布棒在室温下将上述配制剂d33
‑
1至d33
‑
4和d33
‑
c施加至不含油脂的玻璃板上。首先将所得湿膜在室温下干燥30分钟,然后在干燥箱中在60℃下再干燥30分钟。然后,将所得聚合物膜在气候调节室中在黑暗、室温和60%相对湿度下储存如表2所述的时间。在这种情况下,干燥和储存后获得的膜层厚度为40
‑
100μm。
[0271]
根据din en iso 1522测定由所用的配制剂d33
‑
1至d33
‑
4和d33
‑
c获得的聚合物膜在干燥和储存之后的摆阻尼。在这种情况下获得的结果也列于表2中。
[0272]
表2:作为所用羧酸酰肼的函数的摆阻尼的结果
[0273][0274][0275]
从所得结果可以清楚地看出,与没有添加剂的配制剂d33
‑
c相比,所有加入羧酸酰肼的配制剂形成了更硬的聚合物膜。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1