超高极性手性液晶材料、液晶激光器及其制备方法与流程
[0001]
本发明属于液晶材料制备及应用领域,具体涉及超高极性手性液晶材料、液晶激光器及其制备方法。
背景技术:
[0002]
液晶是一种重要的光电材料,在光电显示和空间光调制领域都有极为重要的应用价值。通常,向列相液晶只具有取向序而不具备位置序,虽然单个分子具有永久偶极矩,但指向矢向上和向下的分布机率是一样的,因此整个液晶系统的极性是相消的,不具备铁电特性。
[0003]
2017年,英国约克大学的richard mandle和john goodby合成了一种具有大电偶极子的楔形分子。研究发现,该分子在高温表现为普通向列相,但在低温下(小于133℃)呈现出一种新型的、具有铁电特征的向列相结构,即分子排列产生自发极化,向列相分子偶极矩在空间分布上变得有序,形成具有特定取向的畴。同年,日本riken研究所的hiroya nishikawa也发现了一种具有极高介电常数的极性向列相液晶,该材料还表现出极强的二次谐波响应等特性。目前,这种新型向列相的基础研究尚处于起步阶段,但其极强的介电和非线性光学特征使其具有很高的应用价值。
[0004]
通过往向列相液晶加入手性分子可以得到具有周期性螺旋结构的胆甾相,胆甾相液晶能够选择性反射相同手性的圆偏光,起到类似谐振腔的作用。在1988年kopp等人便利用了胆甾相液晶实现了无镜激射,但早期的液晶激光器需要选择合适的激光染料进行掺杂,胆甾相液晶能在外部激发光的作用下形成激光出射,并且激光出射波长在胆甾相液晶的选择反射谱带边沿位置。随后几十年间,对这类“软激光器”的研究主要集中在改变物理机制、提高激光效率和激光波长调谐等方面。但该技术不可避免的都要用到激光染料掺杂提供增益,发光效率低下,稳定性差和荧光漂白等问题无法得到根本解决,推广应用仍存在很大的局限。
技术实现要素:
[0005]
目前,胆甾相激光器离不开染料增益,本发明利用了一种超高极性手性液晶材料避免该问题。所述超高极性手性液晶具有极强的二次谐波响应特征,无需染料掺杂提供增益,自身即可激发光子,这在胆甾相液晶激光器的应用领域尚处首例。而且相比较传统的非线性晶体倍频激光器,这种无需反射镜的液晶激光器具有谐振腔体积小、激光阈值低、制作简单等特点,在光学领域有更加广泛的应用前景。
[0006]
本发明的目的是通过如下措施来达到:
[0007]
一种超高极性手性液晶材料,将一类手性小分子和极性向列相液晶以一定的质量比均匀混合。
[0008]
进一步的,上述超高极性手性液晶材料质量配比为50-95%的极性向列相液晶和5-50%的手性分子;优选的,超高极性手性液晶材料质量配比为70-95%的极性向列相液晶
和5-30%的手性分子。
[0009]
进一步的,所述的极性向列相液晶包括以下结构式中的一种或几种的组合,
[0010][0011]
r1、r2、r3为1-7个碳原子的烷氧基、烷基、氢基或氟基。
[0012]
进一步的,所述手性小分子化合物结构式:
[0013][0014]
r4、r5、r6、r7和r8为1-7个碳原子的烷基、氢基或氟基。
[0015]
进一步地,所述极性胆甾相液晶螺距可由手性分子浓度配比调节,当手性分子浓度可在5-50%之间时,胆甾相螺距可于0.1-1.0微米之间调节,相对应地,胆甾相选择性反射边沿可在紫外到红外光谱范围内连续切换,实现对应波长的shg增强。
[0016]
进一步地,所述超高极性手性液晶材料在一定的温度范围内,具有ε~104的高介电常数和石英晶体的3-10倍的极强的二次谐波响应特征。
[0017]
一种无荧光分子掺杂胆甾相液晶激光器的制作方法:
[0018]
两片由聚酰亚胺平行摩擦配向的玻璃基板,制成间隔为5-20微米的液晶盒,利用毛细作用将胆甾相混合液晶灌入。
[0019]
进一步地,所述灌入的超高极性手性液晶材料在370~440k下退火处理0.5-2小时,使胆甾相形成稳定的平面织构。
[0020]
由上述制作方法制作的液晶激光器。
[0021]
进一步地,所述液晶激光器的超高极性手性液晶材料的极性胆甾相是平面取向的,在不同的手性掺杂剂浓度具有红外至紫外之间可调的选择反射谱波长带。
[0022]
进一步地,所述液晶激光器的超高极性手性液晶材料的极性胆甾相能对外部激发光形成超强二次谐波响应光,所述外部激发光在极性胆甾相的周期性结构中得到二次谐波信号增幅后,作为激光输出,此技术无需其它染料进行增益。
[0023]
进一步地,所述液晶激光器的超高极性手性液晶材料不仅供应超强二次谐波响应的光,同时也可发射二次谐波以外的高次谐波响应光,所述响应光通过极性胆甾相的周期性结构中得到高次谐波信号增幅后,作为激光或超强高次谐波信号光输出,所述液晶激光器相当于高效率的有机波长转换器件。
[0024]
与现有技术相比,本发明具有如下优点和有益效果:
[0025]
本发明所述的超高极性手性液晶材料具有极强的二次谐波响应,其非线性光学特性能够与晶体中的石英相媲美,这在流体软材料中是十分罕见的。通过改变手性分子掺杂浓度,可以调节分子螺距,实现从紫外到红外全波段的二次谐波增强,该技术与现有的染料掺杂技术相比,避免了光漂白和发光效率底下等问题,大大提高了激光器的稳定性,同时对温度敏感性不高,能在较宽的温度范围内工作。与一般的非线性晶体相比,液晶激光器能够通过手性分子掺杂浓度便利的调节分子螺距,而且具有柔软、易加工和成膜特性,能够实现很多晶体无法应用的工作场景,也更具有成本优势,能够较好的应用于激光倍频调制和二次谐波成像等领域。
附图说明
[0026]
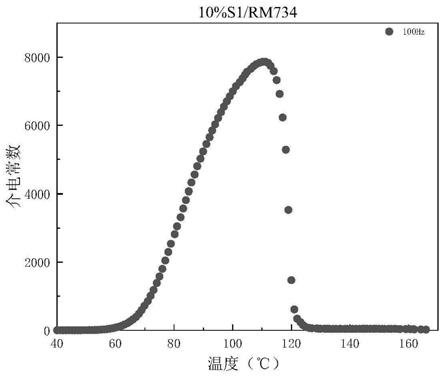
图1是实施例1手性分子浓度为10%掺杂的胆甾相温度介电谱图。
[0027]
图2是实施例1-4手性分子掺杂浓度为5%,10%,20%,30%的dsc图。
[0028]
图3a是实施例1未经退火处理的平面胆甾相的偏光显微照片,有大量的缺陷织构。
[0029]
图3b是实施例2退火后的平面胆甾相的偏光显微照片。
[0030]
图3c是实施例3退火后的平面胆甾相的偏光显微照片。
[0031]
图4是实施例6极性胆甾相激光器示意图。
[0032]
图5是实施例6极性胆甾相激光器二次谐波响应信号随温度变化图。
[0033]
图6是实施例6y-cut石英在室温(25℃)二次谐波强度随旋转角度变化图。
[0034]
图7是实施例2-5掺杂胆甾相的螺距和掺杂分子浓度之间关系图。
[0035]
图8为化合物1的2,4-二甲氧基苯甲酸4-((4-硝基苯氧基)羰基)苯酯的dsc图;
[0036]
图9为化合物1的2,4-二甲氧基苯甲酸4-((4-硝基苯氧基)羰基)苯酯由液相进入向列相时的偏光显微镜(pom)图;
[0037]
图10为化合物1的2,4-二甲氧基苯甲酸4-((4-硝基苯氧基)羰基)苯酯由向列相进入极性向列相(畴区内指向矢方向不同)的偏光显微镜(pom)图;
[0038]
图11为化合物1的2,4-二甲氧基苯甲酸4-((4-硝基苯氧基)羰基)苯酯的介电强度随温度和频率变化的三维曲线图;图中频率(hz)对应的坐标刻度左下至右上分别为106、105、104、103、102、101,温度(℃)对应的坐标刻度分别是100、120、140、160、180、200;图12为化合物1的2,4-二甲氧基苯甲酸4-((4-硝基苯氧基)羰基)苯酯在不同温度下的shg信号强度与石英强度的比值图;
具体实施方式
[0039]
下面结合实施例,对本发明作进一步地详细说明,但不用来限制本发明的范围。
[0040]
本发明所使用的手性分子和极性向列相液晶通过以下方法制得,对应的偶极矩参数如下;未列出的手性分子和极性向列相液晶制备方法相似,参考以下制备方法可以制备得到,并且具有同样的大偶极矩特性。
[0041]
[0042]
[0043][0044]
化合物1
[0045]
2,4-二甲氧基苯甲酸4-((4-硝基苯氧基)羰基)苯酯的制备
[0046][0047]
(1)4-((四氢-2h-吡喃-2-基)氧基)苯甲酸:
[0048]
氮气保护下,将对羟基苯甲酸(2.76g,0.02mol),对甲苯磺酸(1.96g,0.0103mol)和20ml乙醚加入到50ml的单口瓶中,形成悬浮液。冰浴0℃下用注射器逐滴加入3,4-二氢-2h-吡喃(2.8ml,0.0307mol),混合液逐渐恢复到室温搅拌5-6h。此时溶液产生大量沉淀,过滤,用20ml乙醚分多次洗涤,真空干燥得到白色粉末2.89g,产率69.3%;1h nmr(400mhz,chloroform-d)δ8.06(d,j=8.7hz,2h,arh),7.10(d,j=8.6hz,2h,arh),5.53(q,j=
2.8hz,1h,ch),3.86(d,j=21.0hz,1h,ch2),3.63(d,j=11.2hz,1h,ch2),2.07
–
1.50(m,6h,ch2).
[0049]
(2)4-硝基苯基4-((四氢-2h-吡喃-2-基)氧基)苯甲酸酯:
[0050]
氮气保护下,将化合物3(10g,45mmol),1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(10.35g,54mmol),n,n-二甲基氨基吡啶(0.71g,0.54mmol)加入到100ml二氯甲烷中。溶液置于冰浴下搅拌1h,此后逐渐恢复到室温继续搅拌14-24h,通过tlc监测反应。反应完成后,用饱和食盐水洗涤三次,乙酸乙酯萃取。有机相无水硫酸镁干燥,过滤,旋干,粗产物用石油醚/乙酸乙酯=3/1作淋洗剂柱层析提纯,得到白色固体产物12g,产率76.8%。1h nmr(500mhz,chloroform-d)δ8.31(d,j=9.1hz,2h,arh),8.12(dd,j=17.7,8.9hz,2h,arh),7.40(d,j=9.2hz,2h,arh),7.05(dd,j=114.9,8.9hz,2h,arh),5.57(s,1h,ch),4.06
–
3.82(m,1h,ch2),3.61(d,j=55.9hz,1h,ch2),2.03-1.64(s,6h,ch2).
[0051]
(3)4-硝基苯基4-羟基苯甲酸酯:
[0052]
将化合物4(1g,2.9mmol),对甲苯磺酸吡啶盐(72.8mg,0.29mmol),20ml四氢呋喃,20ml甲醇加入到100ml的单口瓶中,混合溶液加热到60℃,继续搅拌6-24h直到tlc检测反应完成。停止反应,降到室温,旋蒸除去多于溶剂然后用乙酸乙酯溶解,去离子水洗涤,有机相用饱和食盐水洗涤,有机相无水硫酸镁干燥,过滤,旋干,粗产物用石油醚/乙酸乙酯=2/1作淋洗剂柱层析提纯,得到白色固体产物0.72g,产率95.1%。1h nmr(400mhz,dmso-d6)δ10.64(s,1h,oh),8.34(d,j=9.1hz,2h,arh),8.02(d,j=8.8hz,2h,arh),7.58(d,j=9.1hz,2h,arh),6.95(d,j=8.8hz,2h,arh).
[0053]
(4)2,4-二甲氧基苯甲酸4-((4-硝基苯氧基)羰基)苯酯:
[0054]
氮气氛围下,将化合物3(2.35g,9.07mmol),购得的2,4-二甲氧基苯甲酸(1.73g,9.52mmol),1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(2.6g,13.6mmol),n,n-二甲基氨基吡啶(110mg,0.91mmol),加入到50ml无水二氯甲烷中,溶液置于冰浴下搅拌1h,此后逐渐恢复到室温继续搅拌14-24h,通过tlc监测反应。反应完成后,用饱和食盐水洗涤三次,乙酸乙酯萃取。有机相无水硫酸镁干燥,过滤,旋干,粗产物用石油醚/二氯甲烷=1/1作淋洗剂柱层析提纯,得到白色固体产物2.86g,产率74.51%。1h nmr(500mhz,chloroform-d)δ8.33(d,j=9.1hz,2h),8.25(d,j=8.7hz,2h),8.10(d,j=8.7hz,1h),7.41(dd,j=19.6,8.9hz,4h),6.62
–
6.52(m,2h),3.92(d,j=18.6hz,6h).
[0055]
如图8所示的dsc,化合物1的液晶分子的降温曲线在120℃附近和80℃附近有两个突起,说明该分子在降温过程中发生两种相转变。结合正交偏光显微镜(pom)在取向好的cell中观察,液晶分子在高温120℃附近开始降温时视野由黑变亮,液晶微观取向发生了变化,开始进入向列相(如图9所示)。当降温到80℃附近时,折射率会发生明显变化,pom下观察到视野明显由黑暗背景变亮,液晶微观取向发生了变化,进入极性向列相(如图10所示)。该液晶分子能够在较宽温度范围内呈现热力学稳定的极性向列型液晶结构。
[0056]
通过测试液晶分子在整个相变温度区间的介电系数,发现在其进入极性相后具有104量级的极高介电强度(如图11所示),同时在此温度范围内该分子的极性液晶相具有很好的shg响应(如图12所示)。
[0057]
化合物2
[0058]
4-甲氧基-2-丙氧基苯甲酸4-((4-硝基苯氧基)羰基)苯酯(3)的制备
[0059][0060]
(1)4-甲氧基-2-丙氧基苯甲酸甲酯:
[0061]
氮气保护下,将购买到的反应物2-羟基-4-甲氧基苯甲酸甲酯(2g,10.98mmol)、碳酸钾(3.03g,21.96mmol)加入30ml dmf中,逐滴注入6-溴丙烷(1.62g,13.17mmol),加热回流反应过夜后,用饱和氯化钠水溶液洗涤3遍,然后用乙酸乙酯萃取,旋干有机层的溶剂后,粗产物用石油醚/乙酸乙酯=5/1作淋洗剂柱层析提纯,得到白色粉状产物2.03g,产率82.46%。
[0062]
(2)4-甲氧基-2-丙氧基苯甲酸:
[0063]
将反应物1(1.5g,6.69mmol)溶于60ml thf/meoh/h2o=1/1/1的混合溶液中,加入koh(1.5g,26.76mmol),混合物加热回流过夜,反应完成后逐渐恢复到室温,加入200ml的水,用1m的盐酸溶液调节ph≈1,后用乙酸乙酯萃取。有机相无水硫酸镁干燥,过滤,旋干,粗产物用石油醚/乙酸乙酯=2/1作淋洗剂柱层析提纯,得到白色固体产物1.35g,产率96.01%。
[0064]
(3)4-甲氧基-2-丙氧基苯甲酸4-((4-硝基苯氧基)羰基)苯酯:
[0065]
氮气氛围下,将化合物2(2g,9.51mmol),4-硝基苯基4-羟基苯甲酸酯(2.35g,9.06mmol),1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(2.6g,13.6mmol),n,n-二甲基氨基吡啶(110mg,0.91mmol),加入到50ml无水二氯甲烷中,溶液置于冰浴下搅拌1h,此后逐渐恢复到室温继续搅拌14-24h,通过tlc监测反应。反应完成后,用饱和食盐水洗涤三次,乙酸乙酯萃取。有机相无水硫酸镁干燥,过滤,旋干,粗产物用石油醚/二氯甲烷=1/1作淋洗剂柱层析提纯,得到白色固体产物3.06g,产率74.81%。1h nmr(500mhz,chloroform-d)δ8.38
–
8.31(m,2h),8.26(d,j=8.7hz,2h),8.06(d,j=8.8hz,1h),7.46
–
7.41(m,2h),7.39(d,j=8.7hz,2h),6.57(dd,j=8.8,2.3hz,1h),6.53(d,j=2.2hz,1h),4.03(t,j=6.4hz,2h),3.89(s,3h),1.88(h,j=7.2hz,2h),1.07(t,j=7.4hz,3h).
[0066]
化合物3
[0067]
4-((4-硝基苯氧基)羰基)苯基4-甲氧基-2-(戊氧基)苯甲酸酯通过类似于化合物2所述的那些方法制备。1h nmr(400mhz,chloroform-d)δ8.33(d,j=9.1hz,2h),8.26(d,j=8.7hz,2h),8.06(d,j=8.7hz,1h),7.41(dd,j=17.2,8.9hz,4h),6.59
–
6.54(m,1h),6.52(d,j=2.2hz,1h),4.06(t,j=6.5hz,2h),3.89(s,3h),1.86(dt,j=14.5,6.6hz,2h),1.49(dt,j=14.7,7.1hz,2h),1.37(dt,j=14.9,7.2hz,2h),0.89(t,j=7.3hz,3h).
[0068]
化合物4
[0069]
4-((4-硝基苯氧基)羰基)苯基4-甲氧基-2-(2-甲氧基乙氧基)苯甲酸酯通过类似于化合物2所述的那些方法制备。1h nmr(400mhz,chloroform-d)δ8.31
–
8.23(m,2h),8.23
–
8.16(m,2h),8.00(d,j=8.8hz,1h),7.42
–
7.29(m,4h),6.53(dd,j=8.8,2.3hz,1h),6.49(d,j=2.3hz,1h),4.21
–
4.11(m,2h),3.82(s,3h),3.79
–
3.70(m,2h),3.37(s,3h).
[0070]
化合物5
[0071]
4-((4-硝基苯氧基)羰基)苯基4-甲氧基-2-(3-甲氧基丙氧基)苯甲酸酯通过类似于化合物2所述的那些方法制备。1h nmr(400mhz,chloroform-d)δ8.32
–
8.23(m,2h),8.23
–
8.16(m,2h),8.00(d,j=8.6hz,1h),7.41
–
7.26(m,4h),6.54
–
6.45(m,2h),4.10(t,j=6.2hz,2h),3.82(s,3h),3.53(t,j=6.1hz,2h),3.25(s,3h),2.04(p,j=6.1hz,2h).
[0072]
化合物6
[0073]
2,4-双(2-甲氧基乙氧基)苯甲酸4-((4-硝基苯氧基)羰基)苯酯通过类似于化合物2所述的那些方法制备。1h nmr(400mhz,chloroform-d)δ8.29
–
8.23(m,2h),8.21
–
8.16(m,2h),7.98(dd,j=8.6,1.9hz,1h),7.37
–
7.29(m,4h),6.53(d,j=8.7hz,2h),4.22
–
4.04(m,4h),3.73(dt,j=9.7,4.6hz,4h),3.38(d,j=14.2hz,6h).
[0074]
特别的其中2,4-双(2-甲氧基乙氧基)苯甲酸甲酯(1)化合物的合成。
[0075][0076]
(1)2,4-双(2-甲氧基乙氧基)苯甲酸甲酯:
[0077]
氮气保护下,将购买到的反应物2,4-二羟基-苯甲酸甲酯(2g,11.89mmol)、碳酸钾(9.86g,71.37mmol)加入50ml dmf中,逐滴注入1-溴-2-甲氧基乙烷(3.64g,26.17mmol),加热回流反应过夜后,用饱和氯化钠水溶液洗涤3遍,然后用乙酸乙酯萃取,旋干有机层的溶剂后,粗产物用石油醚/乙酸乙酯=5/1作淋洗剂柱层析提纯,得到白色粉状产物3.21g,产率94.9%。
[0078]
化合物7
[0079]
4-((4-硝基苯氧基)羰基)苯基(s)-2-(仲丁氧基)-4-甲氧基苯甲酸酯(4)的制备
[0080][0081]
(1)(s)4-甲基苯磺酸仲丁酯:
[0082]
在0℃下,向(r)-丁-2-醇(1g,13.49mmol)和三乙胺(2.82ml,20.24mmol),n,n-二甲基氨基吡啶(164mg,1.349mmol)的dcm(50ml)中的溶液中加入4-甲基苯磺酰氯(p-tsoh)(3.86g,20.24mmol)的二氯甲烷溶液,在20分钟内逐滴加完成。将混合物在室温搅拌过夜后,将反应混合物真空浓缩并将残余物溶于乙酸乙酯。用水和盐水洗涤所得溶液,用mgso4干燥并浓缩。油状残余物通过柱色谱法纯化,产率73%。
[0083]
(2)(s)-2-(仲丁氧基)-4-甲氧基苯甲酸甲酯:
[0084]
氮气氛围下,在圆底烧瓶中装入(1)(1g,4.38mmol),2-羟基-4-甲氧基苯甲酸甲酯(0.96g,5.26mmol),k2co3(1.82g,13.14mmol),ki(70mg,0.44mmol),20ml dmf。并将溶液加热回流直至通过tlc判断反应完成(6-48小时),冷却至室温。水(80ml)加入溶液中,用dcm(3
×
100ml)萃取。有机相用无水mgso4干燥,除去溶剂,并将残余物通过色谱纯化,并在真空烘箱中干燥。产率82%。
[0085]
(3)(s)-2-(仲丁氧基)-4-甲氧基苯甲酸的制备参考化合物2中(2)物质的制备。
[0086]
(4)4-((4-硝基苯氧基)羰基)苯基(s)-2-(仲丁氧基)-4-甲氧基苯甲酸酯的制备参考化合物1中(4)的制备。1h nmr(400mhz,chloroform-d)δ8.37
–
8.30(m,2h),8.29
–
8.22(m,2h),8.04(d,j=8.7hz,1h),7.47
–
7.35(m,4h),6.59
–
6.50(m,2h),4.42(h,j=6.0hz,1h),3.89(s,3h),1.82(ddd,j=13.8,7.5,6.2hz,1h),1.71(dtd,j=13.8,7.3,5.7hz,1h),1.37(d,j=6.1hz,3h),1.01(t,j=7.4hz,3h).
[0087]
化合物8
[0088]
4-((4-硝基苯氧基)羰基)苯基(r)-4-(仲丁氧基)-2-甲氧基苯甲酸酯的制备参考化合物7的制备。
[0089]
化合物9
[0090]
4-((4-硝基苯氧基)羰基)苯基(r)-4-甲氧基-2-(2-甲基丁氧基)苯甲酸酯的制备参考化合物7的制备。1h nmr(400mhz,chloroform-d)δ8.37
–
8.30(m,2h),8.30
–
8.23(m,2h),8.06(d,j=8.7hz,1h),7.47
–
7.35(m,4h),6.56(dd,j=8.8,2.3hz,1h),6.52(d,j=2.3hz,1h),3.96
–
3.82(m,5h),1.99
–
1.88(m,1h),1.67
–
1.59(m,1h),1.36
–
1.28(m,1h),1.06(d,j=6.8hz,3h),0.93(t,j=7.5hz,3h).
[0091]
化合物10
[0092]
4-((4-硝基苯氧基)羰基)苯基(s)-2-甲氧基-4-(2-甲基丁氧基)苯甲酸酯的制备参考化合物7的制备。1h nmr(400mhz,chloroform-d)δ8.37
–
8.30(m,2h),8.28
–
8.22(m,2h),8.08(d,j=8.6hz,1h),7.48
–
7.35(m,4h),6.62
–
6.50(m,2h),4.44(h,j=6.1hz,1h),3.01-3.93(s,5h),1.86
–
1.74(m,1h),1.74
–
1.64(m,1h),1.36(d,j=6.1hz,3h),1.01(t,j=7.5hz,3h).
[0093]
化合物11
[0094]
4-((4-硝基苯氧基)羰基)苯基(s)-4-甲氧基-2-(辛烷-2-基氧基)苯甲酸酯的制备参考化合物7的制备。1h nmr(500mhz,chloroform-d)δ8.37
–
8.31(m,2h),8.29
–
8.23(m,2h),8.08(d,j=8.7hz,1h),7.47
–
7.35(m,4h),6.58
–
6.49(m,2h),4.49(h,j=6.1hz,1h),3.93(s,3h),1.82
–
1.73(m,1h),1.69
–
1.59(m,1h),1.51
–
1.37(m,2h),1.36(d,j=6.0hz,5h),1.30(tdd,j=8.8,5.2,2.5hz,5h),0.94
–
0.85(m,3h).
[0095]
化合物12
[0096]
3',4',5'-三氟-2-甲氧基-[1,1'-联苯]-4-基2,6-二氟-4-(5-丙基-1,3-二恶烷-2-基)苯甲酸酯(4)的制备
[0097][0098]
(1)2-(3,5-二氟苯基)-5-丙基-1,3-二恶烷:
[0099]
氮气环境下,将(5g,42.31mmol)2-丙基丙烷-1,3-二醇,3,5-二氟苯甲醛(5.01g,35.26mmol),2,6-二叔丁基-4-甲基苯酚(bht)(116.5mg,0.53mmol),对甲基苯磺酸(p-tsoh)(3.34g,19.39mmol)于甲苯溶液中回流18-24h,冷却后,饱和食盐水洗涤,乙酸乙酯萃取,旋干溶剂得到无色油状液体9.86g,产率96.2%。
[0100]
(2)2,6-二氟-4-(5-丙基-1,3-二恶烷-2-基)苯甲酸:
[0101]
将(10g,41.28mmol)2-(3,5-二氟苯基)-5-丙基-1,3-二恶烷(1)在氮气氛围中加入到四氢呋喃溶液中,并置于-78℃下,搅拌15min,随后缓慢滴加20.64ml的2m丁基锂的正己烷溶液,半个小时内滴加完成,后继续反应3h,随后氮气环境下投入过量干冰或者通入co2气体鼓泡继续反应1h,最后用1m盐酸溶液调ph≈1,溶液中析出大量白色固体,过滤,用
大量水洗涤,烘干得到产物10.68g,产率90.38%。
[0102]
(3)3',4',5'-三氟-2-甲氧基-[1,1'-联苯]-4-醇:
[0103]
氮气氛围下将(1g,4.93mmol)4-溴-3-甲氧基苯酚,(1.04g,5.91mmol)(3,4,5-三氟苯基)硼酸,(2.04g,14.78mmol)碳酸钾投入到甲苯/异丙醇/水体积比为7/7/3的混合溶液中,随后加入(57mg,0.05mmol)四三苯基膦钯(pd(pph3)4)作为催化剂,回流反应14-20h,反应结束后,加入200ml的水洗涤,用乙酸乙酯萃取后旋干溶剂,层析柱提纯得到无色晶体1.05g,产率83.8%。
[0104]
(4)3',4',5'-三氟-2-甲氧基-[1,1'-联苯]-4-基2,6-二氟-4-(5-丙基-1,3-二恶烷-2-基)苯甲酸酯:
[0105]
氮气氛围下,将4-甲氧基-2-丙氧基苯甲酸(2g,6.99mmol),化合物(1)(1.69g,6.65mmol),1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(1.9g,9.98mmol),n,n-二甲基氨基吡啶(85mg,0.66mmol),加入到50ml无水二氯甲烷中,溶液置于冰浴下搅拌1h,此后逐渐恢复到室温继续搅拌14-24h,通过tlc监测反应。反应完成后,用饱和食盐水洗涤三次,乙酸乙酯萃取。有机相无水硫酸镁干燥,过滤,旋干,粗产物用二氯甲烷/石油醚=2/2作淋洗剂,柱层析提纯,得到白色固体3.09g,产率88.9%。1h nmr(400mhz,chloroform-d)δ7.30(d,j=8.3hz,1h,arh),7.22
–
7.10(m,4h,arh),6.94(dd,j=8.3,2.2hz,1h,arh),6.88(d,j=2.1hz,1h,arh),5.40(s,1h,ch),4.26(dd,j=11.8,4.6hz,2h,ch2),3.85(s,3h),3.54(t,j=11.5hz,2h,ch2),2.23
–
2.02(m,1h,ch),1.53(s,1h,ch),1.39
–
1.29(m,3h,ch3),1.14
–
1.09(m,2h,ch2),0.94(t,j=7.4hz,3h,ch3).
[0106]
化合物13
[0107]
2,3',4',5',6-五氟-[1,1'-联苯]-4-基2,6-二氟-4-(5-丙基-1,3-二恶烷-2-基)苯甲酸酯通过类似于化合物12所述的那些方法制备。1h nmr(400mhz,chloroform-d)δ7.17
–
7.10(m,2h),7.05(ddt,j=8.5,7.4,1.2hz,2h),6.99
–
6.89(m,2h),5.33(s,1h),4.28
–
4.13(m,2h),3.57
–
3.39(m,2h),2.07(tddd,j=11.4,9.2,6.9,4.6hz,1h),1.35
–
1.22(m,2h),1.10
–
0.98(m,2h),0.87(t,j=7.3hz,3h).
[0108]
实施例1
[0109]
手性分子掺杂浓度为10%的极性胆甾相液晶的制备方法如下:
[0110]
使用三氯甲烷为溶剂,分别配制一定质量分数的手性小分子和极性向列相液晶溶液,再按手性分子:极性向列相液晶的质量比为1/9的配制混合物溶液,真空干燥后得到均匀混合物,标记为10%s1/rm734。
[0111][0112]
为所述极性向列相液晶,r1、r2为甲基
[0113][0114]
为所述手性分子,r1、r2为-c6h
13
[0115]
使用偏光显微镜和dsc测试确定胆甾相的相变温度范围,利用cano wedge方法确定手性掺杂剂浓度对螺距的影响。
[0116]
其中,胆甾相液晶选择反射的圆偏振光的反射谱中心波长λc与胆甾相液晶的螺距(p)和液晶平均折射率有关,λc=n*p。
[0117]
实施例2
[0118]
手性分子掺杂浓度为5%的极性胆甾相液晶的制备方法如下:
[0119]
使用三氯甲烷为溶剂,分别配制一定质量分数的手性小分子和极性向列相液晶溶液,再按手性分子:极性向列相液晶的质量比为1/19的配制混合物溶液,真空干燥后得到均匀混合物,标记为5%s1/rm734。
[0120]
实施例3
[0121]
手性分子掺杂浓度为20%的极性胆甾相液晶的制备方法如下:
[0122]
使用三氯甲烷为溶剂,分别配制一定质量分数的手性小分子和极性向列相液晶溶液,再按手性分子:极性向列相液晶的质量比为1/4的配制混合物溶液,真空干燥后得到均匀混合物,标记为20%s1/rm734。
[0123]
实施例4
[0124]
手性分子掺杂浓度为30%的极性胆甾相液晶的制备方法如下:
[0125]
使用三氯甲烷为溶剂,分别配制一定质量分数的手性小分子和极性向列相液晶溶液,再按手性分子:极性向列相液晶的质量比为3/7的配制混合物溶液,真空干燥后得到均匀混合物,标记为30%s1/rm734。
[0126]
实施例5
[0127]
手性分子掺杂浓度为50%的极性胆甾相液晶的制备方法如下:
[0128]
使用三氯甲烷为溶剂,分别配制一定质量分数的手性小分子和极性向列相液晶溶液,再按手性分子:极性向列相液晶的质量比为1/1的配制混合物溶液,真空干燥后得到均匀混合物,标记为50%s1/rm734。
[0129]
图1是实施例1手性分子浓度为10%掺杂的胆甾相温度介电谱图,图1中在120℃相变温度附近介电常数急剧增加,进入极性胆甾相;图2是实施例1-4手性分子掺杂浓度为5%,10%,20%,30%的dsc图,5%掺杂样品在125℃开始进入极性胆甾相,10%掺杂样品在120℃开始进入极性胆甾相,20%掺杂样品在110℃开始进入极性胆甾相,30%掺杂样品在83℃开始进入极性胆甾相。图7是手性分子掺杂浓度为5%,20%,30%,50%的螺距测量图,在5%掺杂样品中螺距为427.5nm,在20%掺杂样品中螺距为613.9nm,在30%掺杂样品中螺距为712.5nm,在50%掺杂样品中螺距为825nm。
[0130]
实施例6
[0131]
激光器的制备如下:
[0132]
准备两片有聚酰亚胺涂附的玻璃基板(1cm2),用天鹅绒布摩擦取向后制备成液晶盒,中间间隔5-20微米。将配置好的胆甾相液晶加热至液相,液晶会在毛细力的作用下被吸入液晶盒,其结构如图4所示,图4为实施例6极性胆甾相激光器示意图,由于极性胆甾相的非线性光学效应,使波长为2λ的入射光转化为波长为λ的光波。在400k下退火处理半小时,使胆甾相形成稳定的平面织构(如图3b、图3c),图3b是实施例2退火后的平面胆甾相的偏光显微照片,在5%手性分子掺杂条件下可以实现在光谱430nm处波长的选择性反射;图3c是
实施例3退火后的平面胆甾相的偏光显微照片,在20%手性分子掺杂条件下可以实现在光谱610nm处波长的反射。未退火的由于冷却过程中缺陷的存在,会在相变过程中形成油性条纹织构(如图3a),这些缺陷会对激光器产生不利的影响。
[0133]
若使用1064nm的脉冲激光作为光源,由于极性胆甾相非线性光学特征,会产生532nm的二次谐波,对应的,改变手性分子掺杂浓度至20%,调节螺距使其对应选择性反射边沿532nm处激射,以达到二次谐波增强的效果。使用光电倍增探测器对出射二次谐波进行探测,与同等光强下石英的二次谐波响应光强进行对比(如图5和图6),从图中可以看出,y轴为二次谐波强度,激光器的二次谐波响应光强明显强于石英(同等条件下石英的shg强度小于3)。
[0134]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制。对于本发明所属技术领域的技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或者替换,都应当是为属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1