制备经表面反应碳酸钙的方法与流程
1.本发明涉及一种用于制备经表面反应碳酸钙的方法、通过所述方法获得的经表面反应碳酸钙及其用途。
背景技术:
2.在实践中,碳酸钙被大量地用在纸、油漆、橡胶和塑料工业中,用于各种目的,例如造纸用涂料、填料、增量剂和颜料以及水性漆和油漆,并且用在水处理中,特别是作为去除无机材料如重金属和/或药物废物如多环化合物、胆固醇和/或内分泌干扰物(edc)的措施。
3.关于阻止碳酸钙粒子聚集并提高这些粒子与添加了所述粒子(例如作为填料或絮凝剂)的物质的亲和力,此类碳酸钙粒子表面的物理和化学性能是通过用脂肪酸或脂肪酸、树脂酸或其他酸的钠盐处理碳酸钙来改变的。
4.在本领域中,已经提出了几种用于改善碳酸钙的化学和物理性能的方法。例如,us 4,219,590a描述了一种用于通过如下方式改善碳酸钙的方法:使碳酸钙粒子与能够与碳酸钙反应的酸性气体进行接触反应,以使粒子尺寸接近精细单分散,同时用酸性气体的酸的钙盐涂覆碳酸钙粒子表面。us 6,666,953 b1涉及一种含有天然碳酸钙的颜料、填料或矿物,其用一种或多种的h3o
+
离子和气态co2的供体处理,允许减少纸的重量以获得恒定的表面积而不会损失物理性能(当其被用作所述纸用的颜料或涂料填料时)。wo 99/02608 a1描述了一种生产耐酸沉淀碳酸钙的高固体浆料的方法,其中固体浆料用化学添加剂如铝酸钠处理,以赋予碳酸钙耐酸性。
5.此外,us 5,584,923a、us 5,647,902 a、us 5,711,799 a、wo 97/08247 a1和wo 98/20079 a1分别描述了碳酸钙,它是耐酸的以能够使其在制造中性至弱酸性纸中用作填料材料;以及用于生产这种耐酸碳酸钙的方法。
6.此外,wo 2005/121257 a2公开了一种用于生产干燥矿物颜料的方法,其特征在于它包含由碳酸钙与下述物质之间的多重反应就地形成的产物:所述碳酸盐与就地形成和/或外部供给的气体co2的一种或多种反应产物;一种或多种通式r
‑
x的化合物。wo 2004/083316 a1涉及无机颜料,它含有通过在碳酸钙和所述碳酸盐与一种或多种中强到强h3o
+
离子供体的一种或多种反应产物,以及所述碳酸盐与就地形成和/或来自外部供应的气体co2的一种或多种反应产物,和至少一种硅酸铝和/或至少一种合成二氧化硅和/或至少一种硅酸钙和/或至少一种硅酸一价盐如硅酸钠和/或硅酸钾和/或硅酸锂优选例如硅酸钠和/或至少一种氢氧化铝和/或至少一种铝酸钠和/或铝酸钾之间的双重和/或多重反应就地形成的产物,其用在造纸应用中,如本体填充和/或纸张涂布。
7.us 5,043,017 a涉及通过如下方式酸稳定化的碳酸钙:将钙螯合剂和共轭碱(例如六偏磷酸钠)之一添加到细碎碳酸钙中,然后加入酸如磷酸。
8.ep 2 264 109 a1涉及一种用于制备表面反应的碳酸钙的方法,其中碳酸钙用至少一种pk
a
小于或等于2.5的酸和至少一种水溶性非聚合物有机和/或无机弱酸和/或其氢盐进行处理,其中该至少一种水溶性非聚合物有机和/或无机弱酸的pk
a
大于2.5,并且其中
其相应的酸阴离子能够形成水不溶性钙盐。wo 2016/113285 a1涉及一种用于生产在ph值为4.5
‑
7的环境中具有改善的稳定性的表面经处理碳酸钙的方法,其中使碳酸钙、至少一种酸(所述酸具有在20℃下测量时为0
‑
8的pk
a
值)和至少一种共轭碱接触以形成表面经处理碳酸钙。
9.ep 3 275 948 a1中描述了一种用于生产经表面反应碳酸钙的方法,其中在水性介质中用磷酸、二氧化碳和特定二羧酸处理含碳酸钙的材料以形成经表面反应碳酸钙的水性悬浮液。
10.但是,本领域仍然需要用于制备高表面积材料的经济方法,以允许提供用于所需目的的特定碳酸钙材料。
11.因此,期望有一种可用的方法,该方法允许制备表面反应的碳酸钙并提供以预定方式改变碳酸钙的比bet表面积和/或孔隙率的可能性。
技术实现要素:
12.因此,本发明的一个目的在于提供一种用于制备经表面反应碳酸钙的经济方法,其中碳酸钙的比bet表面积和/或孔隙率能够以预定方式改变。还期望该方法能够使用标准设备来进行。本发明的另一个目的在于提供一种能够以高收率制备经表面反应碳酸钙的方法。
13.本发明的又一个目的在于提供一种材料,该材料至少部分地源自天然来源并且在环境中不是持久性的,而是易于生物降解的。期望所获得的经表面反应碳酸钙粒子可被用作填料材料,使得它们可以在各种应用中替代常规使用的填料或补充常规使用的填料。还期望经表面反应碳酸钙粒子的官能度可以被控制并且可以针对特定应用进行定制。此外,本发明的一个目的在于提供一种不易受到微生物污染的材料。
14.上述目的以及其他目的由独立权利要求中定义的主题解决。
15.根据本发明的一个方面,提供了一种用于生产经表面反应碳酸钙的方法,包括以下步骤:
16.a)提供含碳酸钙材料,
17.b)提供至少一种无机酸,
18.c)提供至少一种水溶性无机镁盐,和
19.d)在水性介质中用步骤b)的该至少一种无机酸以及二氧化碳处理步骤a)的含碳酸钙材料以形成经表面反应碳酸钙的水性悬浮液,
20.其中该二氧化碳通过无机酸处理就地形成和/或由外部来源供应,并且
21.其中步骤c)的该至少一种水溶性无机镁盐在步骤d)之前、期间和/或之后添加。
22.根据另一方面,提供了能够通过根据本发明的方法获得的经表面反应碳酸钙。
23.根据又一方面,提供了根据本发明的经表面反应碳酸钙在聚合物应用、纸涂层应用、造纸、油漆、涂料、密封剂、印刷油墨、粘合剂、食品、饲料、药物、混凝土、水泥、化妆品、水处理、工程木材应用、石膏板应用、包装应用和/或农业应用中的用途,优选在聚合物和/或食品应用中的用途。
24.根据又一方面,提供了包含根据本发明的经表面反应碳酸钙的制品,其中该制品选自纸产品、工程木材产品、石膏板产品、聚合物产品、卫生产品、医疗产品、保健产品、食品
产品、饲料产品、过滤产品、织造材料、非织造材料、土工织物产品、农业产品、园艺产品、衣物、鞋类产品、行李产品、家用产品、工业产品、包装产品、建筑产品、以及构造产品。
25.本发明的有利实施方案在相应的从属权利要求中定义。
26.根据一种实施方案,该含碳酸钙材料是天然研磨碳酸钙和/或沉淀碳酸钙,优选地,该天然研磨碳酸钙选自大理石、白垩、石灰石及其混合物,和/或该沉淀碳酸钙选自具有文石、球霰石或方解石晶形的沉淀碳酸钙及其混合物。根据进一步的实施方案,该含碳酸钙材料为粒子的形式,该粒子具有0.05
‑
10μm、优选0.2
‑
5.0μm、更优选0.4
‑
3.0μm且最优选0.6
‑
1.2μm的重量中值粒子尺寸d
50
(重量),和/或0.15
‑
55μm、优选1
‑
30μm、更优选2
‑
18μm且最优选3
‑
7μm的重量顶切粒子尺寸d
98
(重量)。
27.根据一种实施方案,该至少一种无机酸选自盐酸、硫酸、亚硫酸、磷酸、其无机酸式盐及其混合物,优选地,该至少一种无机酸选自盐酸、硫酸、亚硫酸、磷酸、由选自nh
4+
、li
+
、na
+
和/或k
+
的阳离子至少部分中和的h2po4‑
、由选自nh
4+
、li
+
、na
+
、k
+
和/或ca
2+
的阳离子至少部分中和的hpo
42
‑
、及其混合物,更优选地,该至少一种无机酸选自盐酸、硫酸、亚硫酸、磷酸或其混合物,并且最优选地,该至少一种无机酸是磷酸。根据进一步的实施方案,该至少一种无机酸以基于该含碳酸钙材料的总重量计为1
‑
60%重量、优选5
‑
55%重量、更优选7
‑
50%重量且最优选10
‑
40%重量的量提供。
28.根据一种实施方案,该至少一种水溶性无机镁盐选自氯化镁、硝酸镁、硫酸镁、硫酸氢镁、溴化镁、碘化镁、氯酸镁、碘酸镁、其水合物及其混合物,优选地,该至少一种水溶性无机镁盐选自溴化镁、硝酸镁、硫酸镁、其水合物及其混合物,并且最优选地,该至少一种水溶性无机镁盐是硫酸镁或其水合物。根据进一步的实施方案,该至少一种水溶性无机镁盐以0.3
‑
270mmol mg
2+
/mol的该含碳酸钙材料的ca
2+
、优选0.7
‑
200mmol mg
2+
/mol的该含碳酸钙材料的ca
2+
、更优选2
‑
135mmol mg
2+
/mol的该含碳酸钙材料的ca
2+
并且最优选3
‑
70mmol mg
2+
/mol的该含碳酸钙材料的ca
2+
的量提供。
29.根据一种实施方案,在步骤d)中,该含碳酸钙材料用包含步骤b)的该至少一种无机酸和步骤c)的该至少一种水溶性无机镁盐的溶液进行处理。根据另一种实施方案,该二氧化碳通过无机酸处理就地形成和/或步骤d)在20
‑
90℃、优选30
‑
85℃、更优选40
‑
80℃、甚至更优选50
‑
75℃并且最优选60
‑
70℃的温度下进行。根据又一种实施方案,该含碳酸钙材料是天然研磨碳酸钙,该至少一种无机酸是磷酸,该至少一种水溶性无机镁盐选自溴化镁、硝酸镁、硫酸镁、其水合物及其混合物,并且优选为硫酸镁或其水合物,并且在步骤d)中,该含碳酸钙材料用包含步骤b)的该至少一种无机酸的和步骤c)的该至少一种水溶性无机镁盐的溶液进行处理。
30.根据一种实施方案,该经表面反应碳酸钙具有使用氮气和bet法测量为20m2/g
‑
200m2/g、优选30m2/g
‑
180m2/g、更优选35m2/g
‑
150m2/g、甚至更优选40m2/g
‑
130m2/g并且最优选50m2/g
‑
100m2/g的比表面积。根据另一种实施方案,该经表面反应碳酸钙具有通过汞孔隙率测定法测量结果计算的粒子内侵入式比孔容为0.1
‑
2.3cm3/g,优选0.2
‑
2.0cm3/g,更优选0.3
‑
1.8cm3/g且最优选0.35
‑
1.6cm3/g,和/或通过汞孔隙率测量确定的粒子内孔尺寸为0.004
‑
1.0μm,优选0.005
‑
0.8μm,更优选0.006
‑
0.6μm且最优选0.007
‑
0.4μm。
31.根据一种实施方案,提供了一种经表面反应碳酸钙,该经表面反应碳酸钙包含含碳酸钙材料,优选方解石;以及至少一种水不溶性钙盐,该水不溶性钙盐选自磷酸三钙和/
或磷灰石磷酸钙,优选选自羟基磷灰石、取代的羟基磷灰石、磷酸八钙及其混合物,更优选选自羟基磷灰石、氟磷灰石、羧基磷灰石及其混合物并且最优选羟基磷灰石,其中该经表面反应碳酸钙包括(i)根据iso 9277:2010使用氮气和bet法测量的比表面积为20
‑
200m2/g;(ii)通过汞孔隙率测定法测量结果计算的粒子内侵入式比孔容为0.1
‑
2.3cm3/g,和/或通过汞孔隙率测量确定的粒子内孔尺寸为0.004
‑
1.0μm,以及(iii)基于该碳酸钙和该至少一种水不溶性钙盐的总量计为至少0.1%重量的量、优选至少0.5%重量的量、更优选至少1%重量的量并且最优选至少2%重量的量的白磷钙石。根据一种实施方案,碳酸钙与该至少一种水不溶性钙盐的质量比在1:0.1至1:76的范围内,优选在1:0.2至1:10的范围内,更优选1:0.5至1:6,甚至更优选1:0.9至1:2,最优选1:0.95至1:1.2。
32.应理解,出于本发明的目的,下列术语具有以下含义:
33.如本文所用的术语“酸”是指在由布朗斯台德
‑
洛瑞(and lowry)定义的含义中的酸(例如h2so4、hso4‑
),其中术语“游离酸”仅指那些处于完全质子化形式的酸(例如h2so4)。
34.术语“水性”悬浮液是指一种体系,其中液相包含水,优选由水组成。然而,所述术语并不排除该水性悬浮液的液相包含少量的至少一种选自甲醇、乙醇、丙酮、乙腈、四氢呋喃及其混合物的水混溶性有机溶剂。如果该水性悬浮液包含至少一种水混溶性有机溶剂,则该水性悬浮液的液相以基于该水性悬浮液的液相的总重量计为0.1
‑
40.0%重量、优选0.1
‑
30.0%重量、更优选0.1
‑
20.0%重量且最优选0.1
‑
10.0%重量的量包含该至少一种水混溶性有机溶剂。例如,该水性悬浮液的液相由水组成。
35.本发明含义中的“含碳酸钙材料”可以是具有基于该含碳酸钙材料的总重量计为至少50%重量、优选75%重量、更优选90%重量并且最优选95%重量的碳酸钙含量的矿物材料或合成材料。
36.除非另外指明,术语“干燥”是指以下这样的过程:根据该过程,至少一部分水从待干燥的材料移除,使得达到在200℃下所获得的“干燥的”材料的恒重。此外,除非另外指明,否则“干燥的”或“干燥”材料可通过其总水分含量来定义,该总水分含量基于该干燥的材料的总重量计为小于或等于1.0%重量、优选小于或等于0.5%重量、更优选小于或等于0.2%重量且最优选0.03
‑
0.07%重量。
37.在本发明含义中的“天然研磨碳酸钙”(gcc)为自天然来源(例如石灰石、大理石或白垩)获得的碳酸钙,并且其通过湿式和/或干式处理如研磨、筛选和/或分级(例如借助于旋风器或分级器)进行加工。
38.除本文的经表面反应碳酸钙之外的颗粒状材料的“粒子尺寸”通过其粒子尺寸的分布d
x
来描述。在其中,值d
x
表示下述这样的直径:相对于该直径,x%重量的粒子具有小于d
x
的直径。这意味着例如d
20
值是指下述这样的粒子尺寸:其中所有粒子的20%重量小于该粒子尺寸。d
50
值因而是重量中值粒子尺寸,也即所有粒子的50%重量大于该粒子尺寸。出于本发明的目的,除非另外指明,否则该粒子尺寸被具体指定为重量中值粒子尺寸d
50
(重量)。粒子尺寸通过使用micromeritics instrument corporation的sedigraph
tm 5100或sedigraph
tm 5120仪器来确定。方法及仪器为本领域技术人员所知且通常用于确定填料和颜料的粒子尺寸。在0.1%重量的na4p2o7的水溶液中进行测量。
39.本文中的经表面反应碳酸钙的“粒子尺寸”被描述为体积基粒子尺寸分布。使用
malvern mastersizer 2000激光衍射系统评价体积中值粒子尺寸d
50
。使用malvern mastersizer 2000激光衍射系统测量的d
50
或者d
98
值所表示的直径值使得以体积计分别为50%或者98%的粒子具有小于该值的直径。使用米氏(mie)理论分析通过测量获得的原始数据,其中粒子折射率为1.57并且吸收系数为0.005。
40.在本发明的上下文中,术语“孔(pore)”被理解为描述在粒子之间和/或在粒子内所发现的空间,也即由粒子在它们在最近相邻接触下包在一起时所形成的空间(粒子间孔),例如在粉末或密实料中,和/或多孔粒子内的空隙空间(粒子内孔),所述空间在被液体饱和时在压力下允许液体通过和/或支持表面润湿液体的吸收。
41.使用micromeritics autopore v 9620汞孔率计,使用汞侵入孔隙率测定法(mercury intrusion porosimetry)测量结果测量比孔容,所述汞孔率计具有最大施加汞压为414mpa(60 000psi),等效于0.004μm(~nm)的拉普拉斯喉径。在每个压力步骤使用的平衡时间是20秒。将样品材料密封在3cm3室的粉末透度计中用于分析。使用软件pore
‑
comp(gane,p.a.c.,kettle,j.p.,matthews,g.p.和ridgway,c.j.,“void space structure of compressible polymer spheres and consolidated calcium carbonate paper
‑
coating formulations”,industrial and engineering chemistry research,35(5),1996年,第1753
‑
1764页),针对汞压缩、透度计膨胀和样品材料压缩来校正数据。
42.在累积侵入数据中见到的总孔体积可被分成两个区域,其中从214μm降至约1
‑
4μm的侵入数据显示具有强烈贡献的任何附聚结构之间的样品的粗填充。在这些直径之下的是粒子自身的精细粒子间填充。如果它们也具有粒子内孔,则此区域显现双峰,并且通过获取由汞侵入比峰转折点更细(即比双峰拐点更细)的孔的比孔容,因而定义比粒子内孔体积。这三个区域的总和给出了粉末的总全部孔体积,但强烈地取决于原始样品压实/在分布的粗孔末端处的粉末的沉降。
43.通过获取累积侵入曲线的第一导数,揭示了基于等效拉普拉斯直径的孔尺寸分布,其必然包括孔屏蔽。微分曲线清楚地显示了粗附聚孔结构区域、粒子间孔区域和粒子内孔区域(如果存在的话)。已知粒子内孔直径范围,则可以从总孔体积中减去剩余粒子间和附聚体间孔体积,以给出在每单位质量孔体积(比孔容)方面的单独的内部孔的希望的孔体积。当然,相同的减法原理也适用于分离任何感兴趣的其他孔尺寸区域。
44.在本发明含义中的“沉淀碳酸钙”(pcc)为合成的物质,通过在水性、半干燥或潮湿环境中在二氧化碳与石灰反应之后沉淀或通过钙和碳酸根离子源在水中沉淀而获得。pcc可以是球霰石、方解石或文石晶型。pcc被描述于例如以下文献中:ep 2 447 213 a1、ep 2 524 898 a1、ep 2 371 766 a1、ep 1 712 597 a1、ep 1 712 523 a1或wo 2013/142473 a1。
45.在本发明含义中的“盐”是由阳离子和阴离子的集合组成的化合物(参见iupac,compendium of chemical terminology,第2版(“gold book”),1997年,“盐(salt)”)。
46.如在整个本文档中所用的材料的“比表面积”(以m2/g表示)可通过利用氮气作为吸附气体的brunauer emmett teller(bet)方法并且通过使用来自micromeritics的asap 2460仪器来确定。该方法为本领域技术人员所熟知并且定义于iso 9277:2010中。测量之前将样品在真空下于100℃调理30分钟的时间周期。通过将该比表面积(以m2/g为单位)与材料的质量(以g为单位)相乘来获得所述材料的总表面积(以m2为单位)。
47.出于本发明的目的,液体组合物的“固体含量”是在所有溶剂或水已经被蒸发之后残余的材料量的量度。如果必要的话,在本发明含义中以%重量给出的悬浮液的“固体含量”可使用来自mettler
‑
toledo的湿度分析仪hr73以5
‑
20g的样品尺寸来确定(t=120℃,自动关闭3,标准干燥)。
48.在本技术含义中的术语“经表面反应(surface
‑
reacted)”将被用来指示材料已经经历了包括在水性环境中部分溶解所述材料的过程,之后是可在不存在或存在另外的结晶添加剂的情况下发生的在所述材料的表面上和围绕所述表面的结晶过程。
49.在本发明含义中的“悬浮液”或“浆料”包括未溶解的固体和水以及任选的其他添加剂,并且通常含有大量固体,并且因而与用于形成其的液体相比更为粘性且可具有更高的密度。
50.出于本发明的目的,术语“粘度”或“布氏粘度”是指布氏粘度。为此目的,通过布氏dv
‑
ii+pro粘度计在25℃
±
1℃下在100rpm下使用布氏rv
‑
转子组的合适转子测量布氏粘度,且具体指定为mpa
·
s。本领域技术人员基于其技术知识将从布氏rv
‑
转子组中选择适合于待测量的粘度范围的转子。例如,对于200
‑
800mpa
·
s之间的粘度范围,可使用转子编号3,对于在400
‑
1600mpa
·
s之间的粘度范围,可使用转子编号4,对于800
‑
3200mpa
·
s之间的粘度范围,可使用转子编号5,对于1000
‑
2 000 000mpa
·
s之间的粘度范围,可使用转子编号6,且对于4000
‑
8 000 000mpa
·
s之间的粘度范围,可使用转子编号7。
51.出于本发明的目的,“水不溶性”材料被定义为下述这样的材料:当100g所述材料与100g去离子水混合且在20℃下在具有0.2μm孔尺寸的过滤器上过滤以回收液体滤液时,该材料在100g所述液体滤液在95
‑
100℃下蒸发之后提供小于或等于1g的回收的固体材料。“水溶性”材料被定义为下述这样的材料:当100g所述材料与100g去离子水混合且在20℃下在具有0.2μm孔尺寸的过滤器上过滤以回收液体滤液时,该材料在100g所述液体滤液在95
‑
100℃下蒸发之后提供大于1g的回收的固体材料。
52.在谈论单数名词时使用不定冠词或定冠词如“a”、“an”或“the”的情况下,这包括了该名词的复数,除非任意情况下另外具体指出。
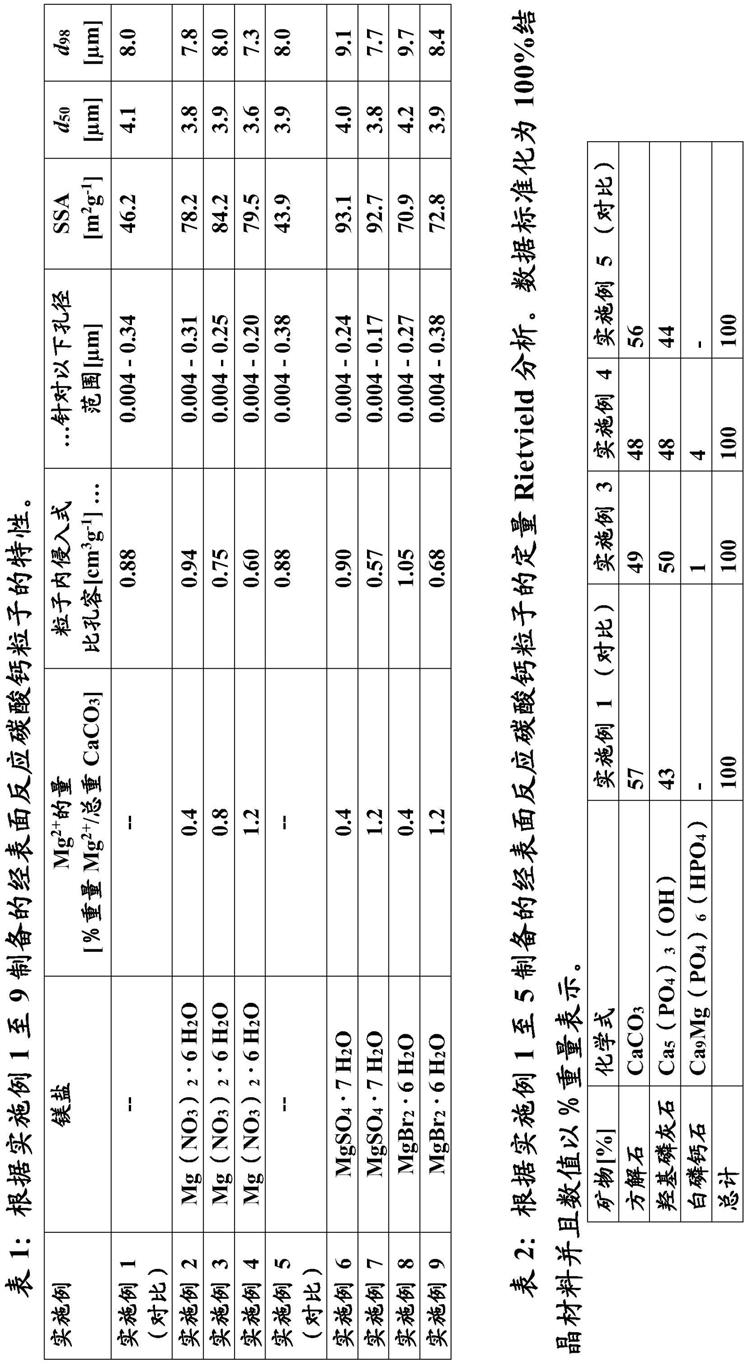
53.当术语“包括或包含(comprising)”在本说明书和权利要求书中被使用时,其并不排除其他要素。出于本发明的目的,术语“由
……
组成(consisting of)”被认为是术语“包括或包含(comprising)”的优选实施方案。如果在下文中定义一个组集(group)包括至少一定数目的实施方案,则这也被理解为公开了一个组集,其优选仅由这些实施方案组成。
54.诸如“可获得(obtainable)”或“可定义(definable)”以及“获得(的)(obtained)”或“定义(的)(defined)”的术语可互换使用。这例如意味着,除非上下文另外明确指出,否则术语“获得(的)”并不意味着指示例如一种实施方案必须通过例如术语“获得(的)”之后的步骤序列来获得,虽然术语“获得(的)”或“定义(的)”总是包括此类限制性理解作为优选实施方案。
55.无论何处使用术语“包括或包含(including)”或者“具有(having)”,这些术语被认为等同于如上定义的“包括或包含(comprising)”。
56.用于生产经表面反应碳酸钙的本发明方法包括以下步骤:a)提供含碳酸钙材料,b)提供至少一种无机酸,c)提供至少一种水溶性无机镁盐,和d)在水性介质中用步骤b)的该至少一种无机酸以及二氧化碳处理步骤a)的含碳酸钙材料以形成经表面反应碳酸钙的
水性悬浮液。该二氧化碳通过无机酸处理就地形成和/或由外部来源供应,并且步骤c)的该至少一种水溶性无机镁盐在步骤d)之前、期间和/或之后添加。
57.下面将更详细地阐述本发明组合物的优选实施方案。应理解,这些实施方案和细节也适用于本发明的产品和用途。
58.方法步骤a)
59.根据本发明方法的步骤a),提供含碳酸钙材料。
60.根据一种实施方案,该至少一种含碳酸钙材料具有基于该含碳酸钙材料的总重量计为至少50%重量、优选75%重量、更优选90%重量并且最优选95%重量的碳酸钙含量。根据另一种实施方案,该至少一种含碳酸钙材料由碳酸钙组成。
61.该含碳酸钙材料可选自天然研磨碳酸钙和/或沉淀碳酸钙。根据本发明的一种实施方案,该天然研磨碳酸钙选自大理石、白垩、石灰石及其混合物,和/或该沉淀碳酸钙选自具有文石、球霰石或方解石晶形的沉淀碳酸钙及其混合物。
[0062]“天然研磨碳酸钙”(gcc)被理解为生产自天然存在形式的碳酸钙,采自沉积岩如石灰石或白垩,或者变质大理石岩,或产自蛋壳、海贝壳或珊瑚。天然研磨碳酸钙的来源可包含其他天然存在的成分如碳酸镁、铝硅酸盐等。已知碳酸钙以三种类型的晶体多晶型物存在:方解石、文石及球霰石。方解石为最常见的晶体多晶型物,其被认为是碳酸钙的最稳定的晶形。文石较为少见,其具有离散或丛集的针状斜方晶晶体结构。球霰石为最罕见的碳酸钙多晶型物,且通常不稳定。天然研磨碳酸钙几乎仅属于方解石型多晶型物,据称其为三方菱面体晶系且代表碳酸钙多晶型物的最稳定形式。
[0063]
通常,天然研磨碳酸钙的研磨可以是干式或湿式研磨步骤并且可例如在使得粉碎主要由使用辅助体冲击产生的条件下,用任何传统研磨装置进行,也即在以下的一种或多种中进行:球磨机、棒磨机、振动研磨机、轧碎机、离心冲击研磨机、立式珠磨机、磨碎机、销棒粉碎机、锤磨机、粉磨机、撕碎机、去块机、切割机(knife cutter)或本领域技术人员已知的其他此类设备。在含碳酸钙的矿物材料包含湿式研磨的含碳酸钙的矿物材料的情况下,研磨步骤可在使得发生自体研磨的条件下和/或通过水平球磨和/或本领域技术人员已知的其他此类方法来进行。由此获得的经湿式加工的研磨的天然碳酸钙可通过众所周知的方法,例如通过絮凝、过滤或强制蒸发(在干燥之前)来洗涤并脱水。后续干燥步骤(必要时)可在单一步骤(例如喷雾干燥)中进行,或者在至少两个步骤中进行。还常见地,这种矿物材料进行选矿步骤(例如浮选、漂白或磁性分离步骤)以去除杂质。
[0064]
根据本发明的一种实施方案,天然研磨碳酸钙(gcc)的来源选自大理石、白垩、石灰石或其混合物,并且优选地,该研磨碳酸钙的来源是大理石。根据本发明的一种实施方案,该gcc通过干式研磨获得。根据本发明的另一种实施方案,该gcc通过湿式研磨和随后的干燥获得。
[0065]
根据本发明的一种实施方案,该碳酸钙包括一种类型的天然研磨碳酸钙。根据本发明的另一种实施方案,该碳酸钙包括两种或更多种类型的选自不同来源的天然研磨碳酸钙的混合物。
[0066]
在本发明含义中的“沉淀碳酸钙”(pcc)为合成材料,通常通过在水性环境中在二氧化碳与氢氧化钙反应之后沉淀或通过将钙离子和碳酸根离子(例如,cacl2及na2co3)从溶液中沉淀出来而获得。生产pcc的其他可能方式为石灰纯碱法,或者solvay法,其中pcc为氨
生产的副产物。沉淀碳酸钙以三种初级晶型存在:方解石、文石和球霰石,且对于这些晶型中的每一晶型而言存在许多不同的多晶型物(晶体惯态)。方解石具有三角结构,该三角结构具有典型的晶体惯态如偏三角面体的(s
‑
pcc)、斜方六面体的(r
‑
pcc)、六角形棱柱的、轴面的、胶体的(c
‑
pcc)、立方的及棱柱的(p
‑
pcc)。文石为正斜方晶结构,该正斜方晶结构具有成对六角形棱晶的典型晶体惯态,以及细长棱柱的、弯曲叶片状的、陡锥状、凿尖晶体、分叉树及珊瑚或蠕虫状的形式的多种分类。球霰石属于六方晶系。所获得的pcc浆料可机械脱水和干燥。
[0067]
根据本发明的一种实施方案,该沉淀碳酸钙是下述这样的沉淀碳酸钙:其优选包含文石、球霰石或方解石矿物晶型,或其混合物。
[0068]
沉淀碳酸钙可在用二氧化碳和至少一种无机酸处理之前通过与用于研磨如上所述的天然研磨碳酸钙的相同方式进行研磨。
[0069]
根据一种优选的实施方案,该含碳酸钙材料是研磨碳酸钙。
[0070]
该含碳酸钙材料可以是粒子的形式,其中至少50%重量的粒子具有小于2μm的直径,优选至少70%重量的粒子具有小于2μm的直径,更优选90%重量的粒子具有小于2μm的直径并且最优选90%重量的粒子具有小于1μm的直径。
[0071]
根据本发明的一种实施方案,该含碳酸钙材料为粒子的形式,该粒子具有0.05
‑
10.0μm、优选0.2
‑
5.0μm、更优选0.4
‑
3.0μm且最优选0.6
‑
1.2μm的重量中值粒子尺寸d
50
。
[0072]
根据本发明的另一种实施方案,该含碳酸钙材料为粒子的形式,该粒子具有0.15
‑
55μm、优选1
‑
30μm、更优选2
‑
18μm且最优选3
‑
7μm的顶切粒子尺寸d
98
。
[0073]
该含碳酸钙材料可干燥使用或以水性悬浮液的形式使用。根据一种优选的实施方案,该含碳酸钙材料为水性悬浮液的形式,该水性悬浮液具有的固体含量为1%重量至90%重量,优选3%重量至60%重量,更优选5%重量至40%重量,并且最优选10%重量至25%重量,基于该水性悬浮液的总重量计。
[0074]
根据本发明的一种优选实施方案,该水性悬浮液由水和该含碳酸钙材料组成。
[0075]
另外可选地,该含碳酸钙材料的水性悬浮液可包含其他添加剂,例如分散剂。合适的分散剂可选自基于例如以下物质的聚羧酸盐的均聚物或共聚物:丙烯酸,甲基丙烯酸,马来酸,富马酸或衣康酸,以及丙烯酰胺,或其混合物。丙烯酸的均聚物或共聚物是特别优选的。这类产品的重均分子量m
w
优选在2 000
‑
15 000g/mol的范围内,其中重均分子量m
w
为3 000
‑
7 000g/mol或3 500
‑
6 000g/mol是特别优选的。根据一种例示的实施方案,该分散剂是重均分子量m
w
为2 000
‑
15 000g/mol、优选3 000
‑
7 000g/mol以及最优选3 500
‑
6 000g/mol的聚丙烯酸钠。
[0076]
根据本发明的一种实施方案,方法步骤a)中提供的该含碳酸钙材料是天然研磨碳酸钙和/或沉淀碳酸钙的水性悬浮液,其固体含量范围为1%重量至90%重量,优选3%重量至60%重量,更优选5%重量至40%重量并且最优选10%重量至25%重量,基于该水性悬浮液的总重量计。
[0077]
方法步骤b)
[0078]
根据本发明方法的步骤b),提供至少一种无机酸。
[0079]
该至少一种无机酸可以是在制备条件下产生h3o
+
离子的任何强无机酸、中强或弱无机酸或其混合物。根据本发明,该至少一种无机酸还可以是在制备条件下产生h3o
+
离子的
酸式盐。
[0080]
根据一种实施方案,该至少一种无机酸是在20℃下pk
a
为0或更低的强无机酸。根据另一种实施方案,该至少一种无机酸是在20℃下pk
a
值为0
‑
2.5的中强无机酸。
[0081]
如果20℃下pk
a
为0或更低,则该至少一种无机酸优选选自硫酸、盐酸或其混合物。如果20℃下pk
a
值为0
‑
2.5,则该至少一种无机酸优选选自亚硫酸、磷酸或其混合物。该至少一种无机酸还可以是无机酸式盐,例如由相应阳离子如nh
4+
、li
+
、na
+
或k
+
至少部分中和的hso4‑
或h2po4‑
,或者由相应阳离子如nh
4+
、li
+
、na
+
和/或k
+
至少部分中和的hpo
42
‑
。该至少一种无机酸还可以是一种或多种酸和一种或多种酸式盐的混合物。
[0082]
根据本发明的一种实施方案,该至少一种无机酸选自盐酸、硫酸、亚硫酸、磷酸、其无机酸式盐及其混合物。优选地,该至少一种无机酸选自盐酸、硫酸、亚硫酸、磷酸、由选自nh
4+
、li
+
、na
+
或k
+
的阳离子至少部分中和的h2po4‑
、由选自nh
4+
、li
+
、na
+
和/或k
+
的阳离子至少部分中和的hpo
42
‑
、及其混合物,更优选地,该至少一种无机酸选自盐酸、硫酸、亚硫酸、磷酸或其混合物,并且最优选地,该至少一种无机酸是磷酸。
[0083]
该至少一种无机酸能够以固体形式或溶液形式提供。根据一种优选的实施方案,该至少一种无机酸以溶液的形式提供。
[0084]
根据一种实施方案,该至少一种无机酸以水溶液的形式提供,该水溶液以基于该水溶液的总重量计为0.1
‑
100%重量的量、优选1
‑
80%重量的量、更优选10
‑
50%重量的量并且最优选20
‑
40%重量的量包含该至少一种无机酸。
[0085]
根据另一种实施方案,该至少一种无机酸以基于该含碳酸钙材料的总重量计为1
‑
60%重量、优选5
‑
55%重量、更优选7
‑
50%重量且最优选10
‑
40%重量的量提供。
[0086]
方法步骤c)
[0087]
根据本发明方法的步骤c),提供至少一种水溶性无机镁盐。
[0088]
该至少一种水溶性无机镁盐可选自本领域技术人员已知的任何水溶性无机镁盐,例如氯化镁、硝酸镁、硫酸镁、硫酸氢镁、溴化镁、碘化镁、氯酸镁或碘酸镁。该水溶性无机镁盐可以是无水盐或水合物盐。如本文使用的“水合物”是含有以一定比例结合的水分子作为晶体的组成部分的无机盐。取决于每化学式单位盐的水分子数,水合物可被指定为一水合物、二水合物、三水合物、四水合物、五水合物、六水合物、七水合物、八水合物、九水合物、十水合物、半水合物等。
[0089]
根据一种实施方案,该至少一种水溶性无机镁盐选自氯化镁、硝酸镁、硫酸镁、硫酸氢镁、溴化镁、碘化镁、氯酸镁、碘酸镁、其水合物,及其混合物,优选地,该至少一种水溶性无机镁盐选自溴化镁、硝酸镁、硫酸镁、其水合物及其混合物,并且最优选地,该至少一种水溶性无机镁盐是硫酸镁或其水合物。
[0090]
根据一种实施方案,氯化镁选自无水氯化镁和/或六水氯化镁(mgcl2·
6h2o)。
[0091]
根据一种实施方案,硝酸镁选自无水硝酸镁、二水硝酸镁(mg(no3)2·
2h2o)、六水合硝酸镁(mg(no3)2·
6h2o)或其混合物。
[0092]
根据一种实施方案,硫酸镁选自无水硫酸镁、一水硫酸镁(mgso4·
h2o)、四水硫酸镁(mgso4·
4h2o)、五水硫酸镁(mgso4·
5h2o)、六水硫酸镁(mgso4·
6h2o)、七水硫酸镁(mgso4·
7h2o)或其混合物。
[0093]
根据一种实施方案,溴化镁选自无水溴化镁和/或六水溴化镁(mgbr2·
6h2o)。
[0094]
根据一种实施方案,碘化镁选自无水碘化镁、六水碘化镁(mgi2·
6h2o)、八水碘化镁(mgi2·
8h2o)或其混合物。
[0095]
根据一种实施方案,氯酸镁选自无水氯酸镁和/或六水氯酸镁(mg(clo3)2·
6h2o)。
[0096]
根据一种实施方式,碘酸镁选自无水碘酸镁和/或四水碘酸镁(mg(io3)2·
4h2o)。
[0097]
根据一种实施方案,该至少一种水溶性无机镁盐选自无水氯化镁、六水氯化镁、无水硝酸镁、二水硝酸镁、六水硝酸镁、无水硫酸镁、一水硫酸镁、四水硫酸镁、五水硫酸镁、六水硫酸镁、七水硫酸镁、硫酸氢镁、无水溴化镁、六水溴化镁、无水碘化镁、六水碘化镁、八水碘化镁、无水氯酸镁、六水氯酸镁、无水碘酸镁、四水碘酸镁及其混合物,优选地,该至少一种水溶性无机镁盐选自无水氯化镁、六水氯化镁、无水硝酸镁、二水硝酸镁、六水硝酸镁、无水硫酸镁、一水硫酸镁、四水硫酸镁、五水硫酸镁、六水硫酸镁、七水硫酸镁及其混合物,更优选地,该至少一种水溶性无机镁盐是无水硝酸镁、二水硝酸镁、六水硝酸镁、无水硫酸镁、一水硫酸镁、四水硫酸镁、五水硫酸镁、六水硫酸镁、七水硫酸镁及其混合物,并且最优选地,该至少一种水溶性无机镁盐为无水硫酸镁、一水硫酸镁、四水硫酸镁、五水硫酸镁、六水硫酸镁、七水硫酸镁或其混合物。
[0098]
根据一种实施方案,该至少一种水溶性无机镁盐以0.3
‑
270mmol mg
2+
/mol的该含碳酸钙材料的ca
2+
、优选0.7
‑
200mmol mg
2+
/mol的该含碳酸钙材料的ca
2+
、更优选2
‑
135mmol mg
2+
/mol的该含碳酸钙材料的ca
2+
并且最优选3
‑
70mmol mg
2+
/mol的该含碳酸钙材料的ca
2+
的量提供。
[0099]
根据一种实施方案,该至少一种水溶性无机镁盐的提供量使得所述镁盐中mg
2+
的量基于该含碳酸钙材料的总重量计为0.05
‑
20%重量,优选0.1
‑
15%重量,更优选0.3
‑
10%重量并且最优选0.4
‑
5%重量。
[0100]
根据另一实施方案,该至少一种水溶性无机镁盐以基于该含碳酸钙材料的总重量计为0.05
‑
40%重量、优选0.1
‑
30%重量、更优选0.3
‑
20%重量并且最优选0.5
‑
10%重量的量提供。
[0101]
该至少一种水溶性无机镁盐能够以溶液形式或作为干燥材料提供。
[0102]
根据一种实施方案,该至少一种水溶性无机镁盐作为干燥材料提供。该干燥材料可以呈粉末、薄片、粒料等形式,并且最优选呈粉末的形式。
[0103]
根据另一种实施方案,该至少一种水溶性无机镁盐以水溶液的形式提供,优选下述这样的水溶液:该水溶液以基于该水溶液的总重量计为0.01
‑
10%重量的量、优选以0.1
‑
8%重量的量、更优选以0.4
‑
5%重量的量并且最优选以0.8
‑
2%重量的量包含该至少一种水溶性无机镁盐。
[0104]
方法步骤d)
[0105]
根据本发明方法的步骤d),在水性介质中用步骤b)的该至少一种无机酸以及二氧化碳处理步骤a)的含碳酸钙材料以形成经表面反应碳酸钙的悬浮液,其中该二氧化碳通过无机酸处理就地形成和/或由外部来源供应,并且其中步骤c)的该至少一种水溶性无机镁盐在步骤d)之前、期间和/或之后添加。
[0106]
该含碳酸钙材料可通过如下方式用该至少一种无机酸进行处理:提供该含碳酸钙材料的水性悬浮液并将该至少一种无机酸添加到所述悬浮液中。可将该至少一种无机酸以浓溶液或更稀溶液的形式添加到悬浮液中。
[0107]
根据一种实施方案,步骤c)的该至少一种水溶性无机镁盐在步骤d)之前添加。换言之,该至少一种水溶性无机镁盐可在第一步中提供,并且随后,步骤d)可通过添加步骤a)的含碳酸钙材料和步骤b)的该至少一种无机酸到该水溶性无机镁盐中来进行。步骤a)的含碳酸钙材料和步骤b)的该至少一种无机酸可以以任何顺序或同时添加。
[0108]
额外地或另外可选地,步骤c)的该至少一种水溶性无机镁盐在步骤d)期间添加。根据一种实施方案,步骤a)的含碳酸钙材料在第一步中提供,步骤c)的水溶性无机镁盐在第二步中添加,并且步骤b)的该至少一种无机酸在第三步中添加。根据另一种实施方案,步骤b)的该至少一种无机酸在第一步中提供,步骤c)的水溶性无机镁盐在第二步中添加,并且步骤a)的含碳酸钙材料在第三步中添加。根据又一实施方案,步骤a)的含碳酸钙材料和步骤c)的水溶性无机镁盐在第一步中提供,并且步骤b)的该至少一种无机酸在第二步中添加。根据又一实施方案,步骤b)的该至少一种无机酸和步骤c)的水溶性无机镁盐在第一步中提供,并且步骤a)的含碳酸钙材料在第二步中添加。根据再一实施方案,同时添加步骤a)的含碳酸钙材料、步骤b)的该至少一种无机酸和步骤c)的水溶性无机镁盐。
[0109]
步骤b)的该至少一种无机酸和步骤c)的该至少一种水溶性无机镁盐可以以单独溶液的形式和/或组合溶液的形式提供。
[0110]
根据一种实施方案,在步骤d)中,用包含步骤b)的该至少一种无机酸和步骤c)的该至少一种水溶性无机镁盐的溶液处理该含碳酸钙材料。根据另一种实施方案,步骤c)的该至少一种水溶性无机镁盐在步骤d)期间添加,并且在步骤d)中,该含碳酸钙材料用包含步骤b)的该至少一种无机酸和步骤c)的该至少一种水溶性无机镁盐的溶液进行处理。
[0111]
根据另一种实施方案,在步骤d)中,该含碳酸钙材料用包含步骤b)的该至少一种无机酸的第一部分的第一溶液并且随后用包含步骤b)的该至少一种无机酸的剩余部分和步骤c)的该至少一种水溶性无机镁盐的第二溶液处理。该第一溶液可包含基于该至少一种无机酸的总量计为小于或等于50%重量、优选小于或等于40%重量、更优选小于或等于30%重量并且最优选小于或等于20%重量的该至少一种无机酸。例如,该第一溶液可包含基于该至少一种无机酸的总量计为0.1
‑
50%重量、优选1
‑
40%重量、更优选5
‑
30%重量并且最优选10
‑
20%重量的该至少一种无机酸。
[0112]
根据又一种实施方案,在步骤b)中提供第一无机酸和第二无机酸,并且在步骤d)中用包含第一无机酸的第一溶液并且随后用包含第二无机酸和步骤c)的该至少一种水溶性无机镁盐的第二溶液处理该含碳酸钙材料。
[0113]
根据一种实施方案,在步骤d)中,该含碳酸钙材料用包含步骤b)的该至少一种无机酸的第一部分的第一溶液并且随后用包含步骤b)的该至少一种无机酸的剩余部分和步骤c)的该至少一种水溶性无机镁盐的第二溶液处理,其中该第一溶液包含基于该至少一种无机酸的总量计为小于50%重量、优选小于40%重量、更优选小于30%重量并且最优选小于20%重量的该至少一种无机酸。
[0114]
额外地或另外可选地,步骤c)的该至少一种水溶性无机镁盐在步骤d)之后添加。换言之,步骤a)的含碳酸钙材料和步骤b)的该至少一种无机酸可在第一步中接触,并且随后可添加该水溶性无机镁盐。步骤a)的含碳酸钙材料和步骤b)的该至少一种无机酸可以以任何顺序或同时接触。
[0115]
根据一种优选的实施方案,在步骤d)中,该含碳酸钙材料用下述溶液处理:该溶液
包含基于该水溶液的总重量计为1
‑
80%重量的量、优选2
‑
50%重量的量、更优选5
‑
40%重量的量并且最优选10
‑
30%重量的量的该至少一种无机酸,以及基于该水溶液的总重量计为0.01
‑
40%重量的量、优选0.1
‑
30%重量的量、更优选0.4
‑
20%重量的量并且最优选0.8
‑
15%重量的量的该至少一种水溶性无机镁盐。
[0116]
根据一种优选的实施方案,该含碳酸钙材料是天然研磨碳酸钙,该至少一种无机酸是磷酸,该至少一种水溶性无机镁盐选自溴化镁、硝酸镁、硫酸镁、其水合物及其混合物,其中步骤c)的该至少一种水溶性无机镁盐在步骤d)期间添加,并且该含碳酸钙材料用包含步骤b)的该至少一种无机酸和步骤c)的该至少一种水溶性无机镁盐的溶液处理。
[0117]
根据一种优选的实施方案,该含碳酸钙材料是天然研磨碳酸钙,该至少一种无机酸是磷酸,该至少一种水溶性无机镁盐选自溴化镁、硝酸镁、硫酸镁、其水合物及其混合物,其中步骤c)的该至少一种水溶性无机镁盐在步骤d)期间添加,并且该含碳酸钙材料用包含步骤b)的该至少一种无机酸的第一部分的第一溶液并且随后用包含步骤b)的该至少一种无机酸的剩余部分和步骤c)的该至少一种水溶性无机镁盐的第二溶液处理。
[0118]
根据一种实施方案,该至少一种无机酸在至少1分钟、优选至少5分钟并且更优选至少10分钟的时间段内添加。在该含碳酸钙材料用包含步骤b)的该至少一种无机酸和步骤c)的该至少一种水溶性无机镁盐的溶液处理的情况下,可以在至少1分钟、优选至少5分钟并且更优选至少10分钟的时间段内添加所述溶液。在该含碳酸钙材料用第一和第二溶液处理的情况下,可以在至少1分钟、优选至少5分钟并且更优选至少10分钟的时间段内添加包含该至少一种无机酸的第一部分或第一无机酸的第一溶液,并且可以在至少1分钟、优选至少5分钟并且更优选至少10分钟的时间段内添加包含该至少一种无机酸的剩余部分或第二无机酸和该至少一种水溶性无机镁盐的第二溶液。
[0119]
根据本发明方法的步骤d),该含碳酸钙材料用二氧化碳处理。取决于无机酸的类型及其浓度,该二氧化碳可在该含碳酸钙材料的无机酸处理过程中自动形成。额外地或另外可选地,可从外部来源供应二氧化碳。
[0120]
无机酸处理和二氧化碳处理可同时进行,这是使用强酸或中强酸时的情况。还可以先进行无机酸处理,例如使用20℃下pk
a
为0
‑
2.5的中强酸,其中二氧化碳就地形成,因此二氧化碳处理将自动与无机酸处理同时进行,然后是用外部来源供应的二氧化碳进行额外处理。
[0121]
根据一种优选的实施方案,二氧化碳通过无机酸处理就地形成。
[0122]
优选地,在步骤d)中形成的悬浮液中气态二氧化碳的浓度(以体积计)使得(悬浮液的体积):(气态co2的体积)的比率为1:0.05至1:70,优选1:0.05至1:60,更优选1:0.05至1:40;并且最优选1:0.05至1:30。
[0123]
在一种优选的实施方案中,步骤d)的无机酸处理步骤和/或二氧化碳处理重复至少一次,更优选多次。
[0124]
在无机酸处理和二氧化碳处理之后,在20℃下测量的水性悬浮液的ph自然达到大于6.0、优选大于6.5、更优选大于7.0的值,从而以ph大于6.0、优选大于6.5、更优选大于7.0的水性悬浮液的形式制备经表面反应的天然或沉淀碳酸钙。
[0125]
根据本发明的一种实施方案,步骤d)在20
‑
90℃、优选30
‑
85℃、更优选40
‑
80℃,甚至更优选50
‑
75℃并且最优选60
‑
70℃的温度下进行。
[0126]
根据一种实施方案,方法步骤d)进行至少1分钟,优选至少5分钟,更优选至少10分钟并且最优选至少15分钟。
[0127]
方法步骤d)可通过简单添加,例如通过倾倒、排出或注入该至少一种无机酸和/或该至少一种水溶性无机镁盐到该含碳酸钙材料中来进行。根据一种实施方案,方法步骤d)在混合条件下进行。合适的混合方法是本领域技术人员已知的。合适的混合方法的实例是摇动、混合、搅拌、搅动、超声处理或通过诸如挡板或薄片的方式引起湍流或层流。合适的混合设备是本领域技术人员已知的,并且可选自例如搅拌器,例如转子定子系统、叶片搅拌器、螺旋桨搅拌器、涡轮搅拌器或锚式搅拌器、静态混合器如包括挡板或薄片的管道。根据本发明的一种示例性实施方案,使用转子定子搅拌器系统。
[0128]
根据另一种示例性实施方案,在步骤d)中,混合所形成的悬浮液以形成基本上层流。本领域技术人员将根据其工艺设备调适混合条件如混合速度和温度。
[0129]
取决于在步骤d)期间通过接触上述化合物引入的水量,可以在方法步骤d)期间引入额外的水,例如为了控制和/或保持和/或实现所得水性悬浮液的所需固体含量或布氏粘度。根据一种实施方案,在步骤d)中获得的混合物的固体含量为1
‑
40%重量,优选5
‑
30%重量,更优选8
‑
25%重量并且最优选10
‑
20%重量,基于该混合物的总重量计。所得水性悬浮液的布氏粘度可为10
‑
10000mpa
·
s,优选50
‑
1000mpa
·
s。
[0130]
本发明的方法可以以连续方法或分批方法的形式进行,优选以分批方法的形式进行。如果合适,方法步骤d)可以重复一次或多次。
[0131]
根据一种实施方案,所得经表面反应碳酸钙浆料的水相可以用去离子水代替。
[0132]
本发明的发明人出人意料地发现,通过本发明的方法,可以以预定的方式增加经表面反应碳酸钙的比bet表面积,例如,与包含碳酸钙的原料相比增加特定百分比。在本发明方法的步骤d)之前、期间和/或之后该至少一种水溶性无机镁盐的添加可导致经表面反应碳酸钙粒子具有增加的比bet表面积。此外还发现,所获得的经表面反应碳酸钙的粒子尺寸没有受到添加该至少一种水溶性无机镁盐的显著影响。因此,本发明的方法可提供独立于粒子尺寸分布而选择性地增加经表面反应碳酸钙的比bet表面积的可能性。本发明人还发现,碳酸钙的孔隙率可以被控制并转移到更高或更低的值。因此,本发明提供了为特定应用定制经表面反应碳酸钙的物理性能的可能性。而且还发现,在本发明的方法中仅使用无机材料避免或至少显著抑制微生物在所得的经表面反应碳酸钙的浆料中的生长。
[0133]
根据一种实施方案,本发明方法导致获得下述这样的经表面反应碳酸钙:该经表面反应碳酸钙具有的比表面积(bet)比通过仅用该至少一种无机酸处理步骤a)中提供的含碳酸钙材料而获得的经表面反应碳酸钙的比表面积(bet)大至少15%、优选大至少40%、更优选大至少60%、甚至更优选大至少100%并且最优选大至少120%,该比表面积使用氮气和bet法测量。例如,本发明方法导致获得下述这样的经表面反应碳酸钙:该经表面反应碳酸钙具有的比表面积(bet)比通过仅用该至少一种无机酸处理步骤a)中提供的含碳酸钙材料而获得的经表面反应碳酸钙的比表面积(bet)大15
‑
39%,优选大40
‑
59%,更优选大60
‑
99%并且最优选大100%
‑
120%,该比表面积使用氮气和bet法测量。
[0134]
该经表面反应碳酸钙可具有通过汞孔隙率测定法测量结果计算的粒子内侵入式比孔容为0.1
‑
2.3cm3/g,优选0.2
‑
2.0cm3/g,更优选0.4
‑
1.8cm3/g且最优选0.6
‑
1.6cm3/g。
[0135]
该经表面反应碳酸钙的粒子内孔尺寸优选为0.004
‑
1.0μm,更优选0.005
‑
0.8μm,
特别优选0.006
‑
0.6μm且最优选0.007
‑
0.4μm,通过汞孔隙率测定法测量结果确定。
[0136]
根据本发明的一种实施方案,该含碳酸钙材料是天然研磨碳酸钙,该至少一种无机酸是磷酸,该至少一种水溶性无机镁盐选自溴化镁、硝酸镁、硫酸镁、其水合物、以及其混合物,并且优选是硫酸镁或其水合物,并且在步骤d)中,该含碳酸钙材料用包含步骤b)的该至少一种无机酸和步骤c)的该至少一种水溶性无机镁盐的溶液处理。优选地,二氧化碳通过无机酸处理就地形成和/或步骤d)在20
‑
90℃、优选30
‑
85℃、更优选40
‑
80℃,甚至更优选50
‑
75℃并且最优选60
‑
70℃的温度下进行。此外,该至少一种无机酸可以以基于该含碳酸钙材料的总重量计为1
‑
60%重量、优选5
‑
55%重量、更优选7
‑
50%重量且最优选10
‑
40%重量的量提供,和/或该至少一种水溶性无机镁盐可以以0.3
‑
270mmol mg
2+
/mol的该含碳酸钙材料的ca
2+
、优选0.7
‑
200mmol mg
2+
/mol的该含碳酸钙材料的ca
2+
、更优选2
‑
135mmol mg
2+
/mol的该含碳酸钙材料的ca
2+
并且最优选3
‑
70mmol mg
2+
/mol的该含碳酸钙材料的ca
2+
的量提供。
[0137]
附加方法步骤
[0138]
根据一种实施方案,本发明的方法进一步包括在步骤d)之后搅拌水性悬浮液的步骤。优选地,将悬浮液搅拌至少1分钟,优选至少5分钟,更优选至少10分钟,并且最优选至少15分钟。
[0139]
该经表面反应碳酸钙的水性悬浮液可进行进一步加工,例如,该经表面反应碳酸钙可与水性悬浮液分离和/或经受干燥步骤。
[0140]
根据一种实施方案,本发明的方法还包括从步骤d)中获得的水性悬浮液中分离该经表面反应碳酸钙的步骤e)。因此,制造经表面反应碳酸钙的方法可包括以下步骤:
[0141]
a)提供含碳酸钙材料,
[0142]
b)提供至少一种无机酸,
[0143]
c)提供至少一种水溶性无机镁盐,和
[0144]
d)在水性介质中用步骤b)的该至少一种无机酸以及二氧化碳处理步骤a)的含碳酸钙材料以形成经表面反应碳酸钙的水性悬浮液,
[0145]
其中该二氧化碳通过无机酸处理就地形成和/或由外部来源供应,并且
[0146]
其中步骤c)的该至少一种水溶性无机镁盐在步骤d)之前、期间和/或之后添加,以及
[0147]
e)从步骤d)中获得的水性悬浮液中分离该经表面反应碳酸钙。
[0148]
可以通过本领域技术人员已知的任何常规分离手段将从步骤d)获得的经表面反应碳酸钙与水性悬浮液分离。根据本发明的一种实施方式,在方法步骤e)中,该经表面反应碳酸钙被机械和/或热分离。机械分离过程的实例是过滤,例如通过转鼓过滤器或压滤机,纳滤或离心。热分离过程的实例是通过例如在蒸发器中施加热的浓缩过程。根据一种优选的实施方案,在方法步骤e)中,机械分离该经表面反应碳酸钙,优选通过过滤和/或离心来进行。
[0149]
在分离之后,可将该经表面反应碳酸钙干燥以获得干燥的经表面反应碳酸钙。根据一种实施方案,本发明的方法进一步包括步骤f):在步骤d)之后或在步骤e)(如果存在的话)之后在60
‑
600℃范围内的温度下干燥该经表面反应碳酸钙,优选直到该经表面反应碳酸钙的水分含量为0.01
‑
5%重量,基于干燥的经表面反应碳酸钙的总重量计。
[0150]
根据本发明的一种实施方案,提供了一种用于制造干燥的经表面反应碳酸钙的方法,包括以下步骤:
[0151]
a)提供含碳酸钙材料,
[0152]
b)提供至少一种无机酸,
[0153]
c)提供至少一种水溶性无机镁盐,和
[0154]
d)在水性介质中用步骤b)的该至少一种无机酸以及二氧化碳处理步骤a)的含碳酸钙材料以形成经表面反应碳酸钙的水性悬浮液,
[0155]
其中该二氧化碳通过无机酸处理就地形成和/或由外部来源供应,并且
[0156]
其中步骤c)的该至少一种水溶性无机镁盐在步骤d)之前、期间和/或之后添加,
[0157]
e)从步骤d)中获得的水性悬浮液中分离该经表面反应碳酸钙,以及
[0158]
f)干燥该经表面反应碳酸钙。
[0159]
通常,干燥步骤f)可以使用任何合适的干燥设备进行并且可以例如包括热干燥和/或在减压下干燥,使用设备如蒸发器、闪蒸干燥器、烘箱、喷雾干燥器和/或在真空室中干燥。干燥步骤f)可以在减压、环境压力或加压下进行。对于低于100℃的温度,可优选在减压下进行该干燥步骤。
[0160]
根据一种优选的实施方案,该分离通过热方法进行。这可以允许随后在不改变设备的情况下干燥该经表面反应碳酸钙。
[0161]
根据一种实施方案,在方法步骤f)中,将该经表面反应碳酸钙干燥,直到形成的经表面反应碳酸钙的水分含量小于或等于1.0%重量,优选小于或等于0.5%重量并且更优选小于或等于0.2%重量,基于该干燥的经表面反应碳酸钙的总重量计。根据另一种实施方案,在方法步骤d)中,该经表面反应碳酸钙被干燥,直到形成的经表面反应碳酸钙的水分含量为0.01
‑
0.15%重量,优选0.02
‑
0.10%重量并且更优选0.03
‑
0.07%重量,基于该干燥的经表面反应碳酸钙的总重量计。
[0162]
根据本发明的一种实施方案,该方法进一步包括步骤g):用至少一种疏水剂处理在步骤d)、e)或f)中获得的经表面反应碳酸钙粒子,以获得下述这样的经表面反应碳酸钙:该经表面反应碳酸钙包含在可及表面区域的至少一部分上的包含该疏水剂的处理层,该疏水剂优选是碳原子总量为c4至c24的脂族羧酸和/或至少一种单取代的琥珀酸酐(其由利用选自在取代基中的碳原子总量为c2至c30的线性、支化、脂族和环状基团的基团单取代的琥珀酸酐组成)和/或一种或多种磷酸单酯和一种或多种磷酸二酯的磷酸酯共混物。
[0163]
应指出,步骤g)独立于步骤e)和/或f)。
[0164]
处理步骤g)中使用的疏水剂可以是本领域技术人员已知的能够在该经表面反应碳酸钙的可及表面区域的至少一部分上形成疏水处理层的任何试剂。
[0165]
用至少一种单取代的琥珀酸酐和/或用至少一种磷酸酯共混物处理在步骤d)、e)或f)中获得的经表面反应碳酸钙的方法步骤g)以及用于涂布的合适化合物被描述于ep 2 722 368 a1和ep 2 770 017 a1中,所述文件因而通过引用并入本文。
[0166]
用于处理在步骤d)、e)或f)中获得的经表面反应碳酸钙粒子的合适脂族羧酸例如是具有4
‑
24个碳原子的脂族线性或支化羧酸并且被描述于ep 3 042 878 a1中。
[0167]
根据一种实施方案,本发明的方法不包括添加选自以下的化合物的步骤:硅酸铝、合成二氧化硅、硅酸钙、一价盐的硅酸盐、铝酸钠、铝酸钾或其混合物。此外或另外可选地,
本发明的方法不包括添加选自以下的化合物的步骤:滑石、高岭土、二氧化钛、氧化镁或其混合物。
[0168]
经表面反应碳酸钙
[0169]
根据本发明的另一方面,提供了经表面反应碳酸钙,其中该经表面反应碳酸钙能够通过本发明的方法获得。因此,该经表面反应碳酸钙可通过包括以下步骤的方法获得:
[0170]
a)提供含碳酸钙材料,
[0171]
b)提供至少一种无机酸,
[0172]
c)提供至少一种水溶性无机镁盐,和
[0173]
d)在水性介质中用步骤b)的该至少一种无机酸以及二氧化碳处理步骤a)的含碳酸钙材料以形成经表面反应碳酸钙的水性悬浮液,其中该二氧化碳通过无机酸处理就地形成和/或由外部来源供应,并且
[0174]
其中步骤c)的该至少一种水溶性无机镁盐在步骤d)之前、期间和/或之后添加。
[0175]
该经表面反应碳酸钙可具有不同的粒子形状,例如玫瑰、高尔夫球和/或大脑的形状。
[0176]
根据一种实施方案,该经表面反应碳酸钙具有使用氮气和bet法测量为20m2/g
‑
200m2/g、优选30m2/g
‑
180m2/g、更优选35m2/g
‑
150m2/g、甚至更优选40m2/g
‑
130m2/g、最优选50m2/g
‑
100m2/g的比表面积。在本发明含义中的bet比表面积被定义为粒子的表面积除以粒子的质量。如本文中所使用,该比表面积使用bet等温线通过吸附测量(iso 9277:2010)且以m2/g具体指定。
[0177]
根据一种实施方式,该经表面反应碳酸钙具有1
‑
75μm、优选2
‑
50μm、更优选2
‑
40μm、甚至更优选2.5
‑
30μm并且最优选3
‑
15μm的体积确定的中值粒子尺寸d
50
(体积),和/或2
‑
150μm、优选4
‑
100μm、更优选4
‑
80μm、甚至更优选5
‑
60μm并且最优选7
‑
30μm的体积确定的顶切粒子尺寸d
98
(体积)。
[0178]
该经表面反应碳酸钙可具有通过汞孔隙率测定法测量结果计算的粒子内侵入式比孔容为0.1
‑
2.3cm3/g,优选0.2
‑
2.0cm3/g,更优选0.3
‑
1.8cm3/g且最优选0.35
‑
1.6cm3/g。
[0179]
该经表面反应碳酸钙的粒子内孔尺寸优选为0.004
‑
1.0μm,更优选0.005
‑
0.8μm,特别优选0.006
‑
0.6μm且最优选0.007
‑
0.4μm,通过汞孔隙率测定法测量结果确定。
[0180]
根据一种实施方案,提供了经表面反应碳酸钙,其中该经表面反应碳酸钙包含含碳酸钙材料、至少一种非碳酸钙的水不溶性钙盐和至少一种水不溶性镁盐。
[0181]
根据一种实施方案,提供了经表面反应碳酸钙,其中该经表面反应碳酸钙包含含碳酸钙材料和至少一种非碳酸钙的水不溶性钙盐,例如磷酸三钙和/或磷灰石磷酸钙,优选羟基磷灰石、磷酸八钙、氟磷灰石、羧基磷灰石或其混合物。该经表面反应碳酸钙可包含碳酸钙与磷酸三钙和/或磷灰石磷酸钙的质量比,其范围为0.01:1至59:1,优选0.1:1至44:1,更优选0.2:1至29:1,甚至更优选0.3:1至15:1并且最优选0.5:1至5:1。
[0182]
根据一种实施方案,该经表面反应碳酸钙包含含碳酸钙材料和至少一种水不溶性钙盐,该水不溶性钙盐选自磷酸三钙和/或磷灰石磷酸钙,优选选自羟基磷灰石、取代的羟基磷灰石、磷酸八钙及其混合物。羟基磷灰石可以以未取代或取代的形式存在。取代的羟基磷灰石的例子是氟磷灰石或羧基磷灰石。根据一种实施方案,该水不溶性钙盐选自羟基磷灰石、氟磷灰石、羧基磷灰石及其混合物,并且最优选羟基磷灰石。根据一种实施方案,该含
碳酸钙材料是方解石。该经表面反应碳酸钙可包含碳酸钙与该水不溶性钙盐的质量比,其范围为1:0.1至1:76,优选1:0.2至1:10,更优选1:0.5至1:6,甚至更优选1:0.9至1:2,最优选1:0.95至1:1.2。根据一种优选实施方案,该含碳酸钙材料为方解石,该至少一种水不溶性钙盐为羟基磷灰石,且方解石与羟基磷灰石的质量比的范围为1:0.1至1:76,优选1:0.2至1:10,更优选1:0.5至1:6,甚至更优选1:0.9至1:2,最优选1:0.95至1:1.2。
[0183]
本领域技术人员将会理解,碳酸钙与水不溶性钙盐的质量比可以通过该至少一种无机酸的添加量来控制。例如,以基于含碳酸钙材料的总重量计提供5%重量的量的磷酸可导致碳酸钙与羟基磷灰石的质量比为约1:0.1。根据一种示例性实施方案,该含碳酸钙材料是天然研磨碳酸钙和/或沉淀碳酸钙,该至少一种无机酸是磷酸并且以基于含碳酸钙材料的总重量计为10%重量的量提供,该至少一种水不溶性钙盐为羟基磷灰石,并且碳酸钙与羟基磷灰石的质量比为1:0.18至1:0.22。根据另一示例性实施方案,该含碳酸钙材料是天然研磨碳酸钙和/或沉淀碳酸钙,该至少一种无机酸是磷酸并且以基于含碳酸钙材料的总重量计为20%重量的量提供,该至少一种水不溶性钙盐为羟基磷灰石,并且碳酸钙与羟基磷灰石的质量比为1:0.48至1:0.54。根据又一示例性实施方案,该含碳酸钙材料是天然研磨碳酸钙和/或沉淀碳酸钙,该至少一种无机酸是磷酸并且以基于含碳酸钙材料的总重量计为30%重量的量提供,该至少一种水不溶性钙盐为羟基磷灰石,并且碳酸钙与羟基磷灰石的质量比为1:0.95至1:1.05。根据再一示例性实施方案,该含碳酸钙材料是天然研磨碳酸钙和/或沉淀碳酸钙,该至少一种无机酸是磷酸并且以基于含碳酸钙材料的总重量计为50%重量的量提供,该至少一种水不溶性钙盐为羟基磷灰石,并且碳酸钙与羟基磷灰石的质量比为1:5至1:6。
[0184]
本发明的发明人出人意料地发现,与常规生产的经表面反应碳酸钙相比,本发明的经表面反应碳酸钙中结晶水不溶性钙盐的量可以增加。例如已知发现,由磷酸生产的经表面反应碳酸钙可比常规生产的经表面反应碳酸钙含有更多的结晶磷酸三钙和/或磷灰石磷酸钙。不受任何理论的束缚,本发明人相信,至少一种水溶性无机镁盐的添加促进了磷酸钙如羟基磷灰石的结晶,留下较少的无定形磷酸钙。
[0185]
根据本发明的一种实施方案,该经表面反应碳酸钙具有使用氮气和bet法测量为20m2/g
‑
200m2/g、优选30m2/g
‑
180m2/g、更优选35m2/g
‑
150m2/g、甚至更优选40m2/g
‑
130m2/g并且最优选50m2/g
‑
100m2/g的比表面积(bet),并且该经表面反应钙盐粒子包含碳酸钙与磷灰石磷酸钙、优选羟基磷灰石、磷酸八钙、氟磷灰石、羧基磷灰石或其混合物、更优选羟基磷灰石的质量比,其范围为0.01:1至59:1,优选0.1:1至44:1,更优选0.2:1至29:1,甚至更优选0.3:1至15:1,并且最优选0.5:1至5:1。
[0186]
根据本发明的另一种实施方案,该经表面反应碳酸钙具有使用氮气和bet法测量为20m2/g
‑
200m2/g、优选30m2/g
‑
180m2/g、更优选35m2/g
‑
150m2/g、甚至更优选40m2/g
‑
130m2/g并且最优选50m2/g
‑
100m2/g的比表面积(bet),并且该经表面反应钙盐粒子包含碳酸钙与磷灰石磷酸钙、优选羟基磷灰石、取代的羟基磷灰石、磷酸八钙及其混合物、更优选羟基磷灰石、氟磷灰石、羧基磷灰石及其混合物、最优选羟基磷灰石的质量比,其范围为1:0.1至1:76,优选1:0.2至1:10,更优选1:0.5至1:6,甚至更优选1:0.9至1:2,最优选1:0.95至1:1.2。
[0187]
根据本发明的一种实施方案,该至少一种水不溶性镁盐包含白磷钙石(whitlockite)。白磷钙石是一种具有化学式ca9mg(po4)6(hpo4)的矿物,其可以在本发明的
方法期间在该含碳酸钙材料的表面上形成。该经表面反应碳酸钙可进一步包含基于该碳酸钙和该至少一种水不溶性钙盐的总量计为至少0.1%重量的量的白磷钙石,优选的量为至少0.5%重量,更优选的量为至少1%重量,并且最优选的量为至少2%重量。
[0188]
根据本发明的一种实施方案,提供经表面反应碳酸钙,其包含
[0189]
含碳酸钙材料,优选方解石,以及至少一种水不溶性钙盐,选自磷酸三钙和/或磷灰石磷酸钙,优选选自羟基磷灰石、磷酸八钙、氟磷灰石、羧基磷灰石及其混合物,最优选羟基磷灰石,
[0190]
其中该经表面反应碳酸钙包括
[0191]
(i)根据iso 9277:2010使用氮气和bet法测量的比表面积为20
‑
200m2/g;
[0192]
(ii)通过汞孔隙率测定法测量结果计算的粒子内侵入式比孔容为0.1
‑
2.3cm3/g,和/或
[0193]
通过汞孔隙率测量确定的粒子内孔尺寸为0.004
‑
1.0μm,以及
[0194]
(iii)基于该碳酸钙和该至少一种水不溶性钙盐的总量计为至少0.1%重量的量、优选至少0.5%重量的量、更优选至少1%重量的量并且最优选至少2%重量的量的白磷钙石。根据一种实施方案,碳酸钙与该至少一种水不溶性钙盐的质量比在1:0.1至1:76的范围内,优选在1:0.2至1:10的范围内,更优选1:0.5至1:6,甚至更优选1:0.9至1:2,最优选1:0.95至1:1.2。
[0195]
根据本发明的一种优选实施方案,提供包含方解石和羟基磷灰石的经表面反应碳酸钙,
[0196]
其中该经表面反应碳酸钙包括
[0197]
(i)根据iso 9277:2010使用氮气和bet法测量的比表面积为20
‑
200m2/g;
[0198]
(ii)通过汞孔隙率测定法测量结果计算的粒子内侵入式比孔容为0.1
‑
2.3cm3/g,和/或
[0199]
通过汞孔隙率测量确定的粒子内孔尺寸为0.004
‑
1.0μm,以及
[0200]
(iii)基于方解石和羟基磷灰石的总量计为至少0.1%重量的量、优选至少0.5%重量的量、更优选至少1%重量的量并且最优选至少2%重量的量的白磷钙石,
[0201]
其中方解石与羟基磷灰石的质量比在1:0.1至1:76的范围内,优选在1:0.2至1:10的范围内,更优选1:0.5至1:6,甚至更优选1:0.9至1:2,最优选1:0.95至1:1.2。
[0202]
能够通过本发明的方法获得的经表面反应碳酸钙可以以经表面反应碳酸钙的悬浮液、分离的经表面反应碳酸钙或干燥的经表面反应碳酸钙的形式提供。根据一种优选的实施方案,该经表面反应碳酸钙是干燥的经表面反应碳酸钙。
[0203]
在该经表面反应碳酸钙已被干燥的情况下,该干燥的经表面反应碳酸钙的水分含量基于该干燥的经表面反应碳酸钙的总重量计可以为0.01
‑
5%重量。根据一种实施方案,该干燥的经表面反应碳酸钙的水分含量基于该干燥的经表面反应碳酸钙的总重量计为小于或等于1.0%重量,优选小于或等于0.5%重量,更优选小于或等于0.2%重量。根据另一种实施方案,该干燥的经表面反应碳酸钙的水分含量基于该干燥的经表面反应碳酸钙的总重量计为0.01
‑
0.15%重量,优选0.02
‑
0.10%重量,更优选0.03
‑
0.07%重量。
[0204]
本发明的经表面反应碳酸钙也可以以组合物的形式提供和/或使用。根据本发明的一个方面,提供包含根据本发明的经表面反应碳酸钙的组合物。此外,可以存在其他填料
材料,例如天然研磨碳酸钙、沉淀碳酸钙、白云石及其混合物。该组合物可包含基于该组合物的总重量计为至少20%重量、优选至少40%重量、更优选至少60%重量且最优选至少80%重量的量的根据本发明的经表面反应碳酸钙。
[0205]
该经表面反应碳酸钙可用于各种应用。
[0206]
根据一种实施方案,能够通过根据本发明的方法获得的经表面反应碳酸钙被用在以下项中:聚合物应用、纸涂层应用、造纸、油漆、涂料、密封剂、印刷油墨、粘合剂、食品、饲料、药物、混凝土、水泥、化妆品、水处理、工程木材应用、石膏板应用、包装应用和/或农业应用。优选地,能够通过根据本发明的方法获得的经表面反应碳酸钙被用在聚合物应用和/或食品应用中。根据一种实施方案,该经表面反应碳酸钙以干燥的经表面反应碳酸钙的形式使用。
[0207]
根据另一方面,提供包含根据本发明的经表面反应碳酸钙的制品,其中该制品选自纸产品、工程木材产品、石膏板产品、聚合物产品、卫生产品、医疗产品、保健产品、过滤产品、织造材料、非织造材料、土工织物产品、农业产品、园艺产品、衣物、鞋类产品、行李产品、家用产品、工业产品、包装产品、建筑产品、以及构造产品。
具体实施方式
[0208]
基于以下实施例将更好地理解本发明的范围和益处,这些实施例旨在说明本发明的某些实施方案并且是非限制性的。
[0209]
实施例
[0210]
1、测量方法
[0211]
下面描述在实施例中实施的测量方法。
[0212]
粒子尺寸分布
[0213]
使用malvern mastersizer 2000 laser diffraction system(malvern instruments plc.,英国)评价体积确定中值粒子尺寸d
50
(体积)和体积确定顶切粒子尺寸d
98
(体积)。d
50
(体积)或d
98
(体积)值指示出的直径值为:分别50%或98%体积的粒子具有小于这个值的直径。使用米氏(mie)理论分析通过测量结果获得的原始数据,其中粒子折射率为1.57并且吸收系数为0.005。方法及仪器为本领域技术人员所知并且通常用于确定填料和颜料的粒子尺寸分布。
[0214]
重量确定的中值粒子尺寸d
50
(重量)和重量确定的顶切粒子尺寸d
98
(重量)通过沉降法测量,所述沉降法是在重力场中的沉降行为的分析。使用美国micromeritics instrument corporation的sedigraph
tm 5120进行测量。方法及仪器为本领域技术人员所知且通常用于确定填料和颜料的颗粒尺寸。在0.1%重量na4p2o7的水溶液中进行测量。使用高速搅拌器及超声处理来分散样品。
[0215]
比表面积(ssa)
[0216]
通过将样品在250℃下加热30分钟的时间周期进行调理之后,通过使用氮气根据iso 9277:2010经由bet方法测量比表面积。在这种测量之前,样品在布氏漏斗内过滤,用去离子水冲洗并且在烘箱中于90
‑
100℃下干燥过夜。随后,将干燥饼在研钵中充分研磨,且将所得粉末在130℃下在湿度平衡下放置,直到达到恒重为止。
[0217]
粒子内侵入式比孔容(以cm3/g表示)
[0218]
使用micromeritics autopore v 9620汞孔率计,使用汞侵入孔隙率测定法测量结果测量比孔容,所述汞孔率计具有最大施加汞压为414mpa(60 000psi),等效于0.004μm(~nm)的拉普拉斯喉径。在每个压力步骤使用的平衡时间是20秒。将样品材料密封在5cm3室的粉末透度计中用于分析。使用软件pore
‑
comp(gane,p.a.c.,kettle,j.p.,matthews,g.p.和ridgway,c.j.,“void space structure of compressible polymer spheres and consolidated calcium carbonate paper
‑
coating formulations”,industrial and engineering chemistry research,35(5),1996年,第1753
‑
1764页),针对汞压缩、透度计膨胀和样品材料压缩来校正数据。
[0219]
在累积侵入数据中见到的总孔体积可被分成两个区域,其中从214μm降至约1
‑
4μm的侵入数据显示具有强烈贡献的任何附聚结构之间的样品的粗填充。在这些直径之下的是粒子自身的精细粒子间填充。如果它们也具有粒子内孔,则此区域显现双峰,并且通过获取由汞侵入比峰转折点更细(即比双峰拐点更细)的孔的比孔容,定义比粒子内孔体积。这三个区域的总和给出了粉末的总全部孔体积,但强烈地取决于原始样品压实/在分布的粗孔末端处的粉末的沉降。
[0220]
通过获取累积侵入曲线的第一导数,揭示了基于等效拉普拉斯直径的孔尺寸分布,其必然包括孔屏蔽。微分曲线清楚地显示了粗附聚孔结构区域、粒子间孔区域和粒子内孔区域(如果存在的话)。已知粒子内孔直径范围,则可以从总孔体积中减去剩余粒子间和附聚体间孔体积,以给出在每单位质量孔体积(比孔容)方面的单独的内部孔的希望的孔体积。当然,相同的减法原理也适用于分离任何感兴趣的其他孔尺寸区域。
[0221]
x射线衍射(xrd)分析
[0222]
使用bruker d8 advance粉末衍射仪遵循布拉格定律分析所制备的样品。此衍射仪由2.2kw x
‑
射线管、样品支架、θ
‑
θ测角仪和检测器构成。在所有实验中采用镍过滤的cu kα辐射。特征图(profiles)是在2θ下使用0.7
°
/分钟的扫描速度自动记录的图(xrd gv 7600)。基于icdd pdf 2数据库的参考图样(xrd ltm 7603)使用diffrac
suite
软件包eva和search通过矿物含量对所得到的粉末衍射图样进行分类。
[0223]
衍射数据的定量分析(即确定多相样品中不同相的量)使用diffrac
suite
软件包topas(xrd ltm_7604)来进行。这涉及对全衍射图样进行建模(rietveld方案),使得经计算的图样重复实验的图样。
[0224]
用以估计大致矿物浓度的半定量(sq)计算使用diffrac
suite
软件包eva进行。半定量分析考虑了图样相对高度和i/icor值(i/icor:目标化合物中最强线的强度与刚玉最强线的强度之比,两者均由50
‑
50重量混合物进行的扫描来测量)来进行。
[0225]
2、实施例
[0226]
实施例1(对比实施例)
[0227]
经表面反应碳酸钙通过如下方式获得:通过调节来自挪威hustadmarmor的研磨大理石碳酸钙的固含量以使得获得基于水性悬浮液的总重量计为20%重量的固含量,在混合容器中制备10升研磨碳酸钙的水性悬浮液。该研磨碳酸钙具有通过沉降测定的90%小于2μm的重量基粒子尺寸分布,d
50
(重量)为0.7μm且d
98
(重量)为3.4μm。
[0228]
另外,将磷酸稀释以使其包含基于溶液总重量计为30%的磷酸。
[0229]
在混合浆料的同时,在10分钟内添加2.3kg的磷酸溶液。在整个实验的过程中,悬
浮液的温度保持在70℃
±
1℃。最后,在添加酸之后,将悬浮液再搅拌5分钟,然后将其从容器中取出并使其冷却。
[0230]
所获得的经表面反应碳酸钙的体积确定的中值粒子尺寸d
50
为4.1μm,体积确定的顶切粒子尺寸d
98
为8.0μm,并且比表面积ssa为46.2m2g
‑1。
[0231]
实施例2
[0232]
经表面反应碳酸钙通过如下方式获得:通过调节来自挪威hustadmarmor的研磨大理石碳酸钙的固含量以使得获得基于水性悬浮液的总重量计为20%重量的固含量,在混合容器中制备10升研磨碳酸钙的水性悬浮液。该研磨碳酸钙具有通过沉降测定的90%小于2μm的重量基粒子尺寸分布,d
50
(重量)为0.7μm且d
98
(重量)为3.4μm。
[0233]
另外,将磷酸稀释以使其包含基于溶液总重量计为30%的磷酸。然后将基于纯磷酸总重量计为14%重量的mg(no3)2·
6h2o添加到该溶液中并搅拌该溶液直至其完全溶解。
[0234]
在混合浆料的同时,在10分钟内添加2.4kg的磷酸溶液。在整个实验的过程中,悬浮液的温度保持在70℃
±
1℃。最后,在添加酸之后,将悬浮液再搅拌5分钟,然后将其从容器中取出并使其冷却。
[0235]
所获得的经表面反应碳酸钙的体积确定的中值粒子尺寸d
50
为3.8μm,体积确定的顶切粒子尺寸d
98
为7.8μm,并且比表面积ssa为78.2m2g
‑1。
[0236]
实施例3
[0237]
经表面反应碳酸钙通过如下方式获得:通过调节来自挪威hustadmarmor的研磨大理石碳酸钙的固含量以使得获得基于水性悬浮液的总质量计为20%重量的固含量,在混合容器中制备10升研磨碳酸钙的水性悬浮液。该研磨碳酸钙具有通过沉降测定的90%小于2μm的重量基粒子尺寸分布,d
50
(重量)为0.7μm且d
98
(重量)为3.4μm。
[0238]
另外,将磷酸稀释以使其包含基于溶液总重量计为30%的磷酸。然后将基于纯磷酸总重量计为28.7%重量的mg(no3)2·
6h2o添加到该溶液中并搅拌该溶液直至其完全溶解。
[0239]
在混合浆料的同时,在10分钟内添加2.5kg的磷酸溶液。在整个实验的过程中,悬浮液的温度保持在70℃
±
1℃。最后,在添加酸之后,将悬浮液再搅拌5分钟,然后将其从容器中取出并使其冷却。
[0240]
所获得的经表面反应碳酸钙的体积确定的中值粒子尺寸d
50
为3.9μm,体积确定的顶切粒子尺寸d
98
为8.0μm,并且比表面积ssa为84.2m2g
‑1。
[0241]
实施例4
[0242]
经表面反应碳酸钙通过如下方式获得:通过调节来自挪威hustadmarmor的研磨大理石碳酸钙的固含量以使得获得基于水性悬浮液的总重量计为20%重量的固含量,在混合容器中制备10升研磨碳酸钙的水性悬浮液。该研磨碳酸钙具有通过沉降测定的90%小于2μm的重量基粒子尺寸分布,d
50
(重量)为0.7μm且d
98
(重量)为3.4μm。
[0243]
另外,将磷酸稀释以使其包含基于溶液总重量计为30%的磷酸。然后将基于纯磷酸总重量计为43.0%重量的mg(no3)2·
6h2o添加到该溶液中并搅拌该溶液直至其完全溶解。
[0244]
在混合浆料的同时,在10分钟内添加2.6kg的磷酸溶液。在整个实验的过程中,悬浮液的温度保持在70℃
±
1℃。最后,在添加酸之后,将悬浮液再搅拌5分钟,然后将其从容
器中取出并使其冷却。
[0245]
所获得的经表面反应碳酸钙的体积确定的中值粒子尺寸d
50
为3.6μm,体积确定的顶切粒子尺寸d
98
为7.3μm,并且比表面积ssa为79.5m2g
‑1。
[0246]
实施例5(对比实施例)
[0247]
经表面反应碳酸钙通过如下方式获得:通过调节来自挪威hustadmarmor的研磨大理石碳酸钙的固含量以使得获得基于水性悬浮液的总重量计为20%重量的固含量,在混合容器中制备10升研磨碳酸钙的水性悬浮液。该研磨碳酸钙具有通过沉降测定的90%小于2μm的重量基粒子尺寸分布,d
50
(重量)为0.7μm且d
98
(重量)为3.4μm。
[0248]
另外,将磷酸稀释以使其包含基于溶液总重量计为30%的磷酸。
[0249]
在混合浆料的同时,在10分钟内添加2.3kg的磷酸溶液。在整个实验的过程中,悬浮液的温度保持在70℃
±
1℃。最后,在添加酸之后,将悬浮液再搅拌5分钟,然后将其从容器中取出并使其冷却。
[0250]
所获得的经表面反应碳酸钙的体积确定的中值粒子尺寸d
50
为3.9μm,体积确定的顶切粒子尺寸d
98
为8.0μm,并且比表面积ssa为43.9m2g
‑1。
[0251]
实施例6
[0252]
经表面反应碳酸钙通过如下方式获得:通过调节来自挪威hustadmarmor的研磨大理石碳酸钙的固含量以使得获得基于水性悬浮液的总重量计为20%重量的固含量,在混合容器中制备10升研磨碳酸钙的水性悬浮液。该研磨碳酸钙具有通过沉降测定的90%小于2μm的重量基粒子尺寸分布,d
50
(重量)为0.7μm且d
98
(重量)为3.4μm。
[0253]
另外,将磷酸稀释以使其包含基于溶液总重量计为30%的磷酸。然后将基于纯磷酸总重量计为4.5%重量的mgso4·
7h2o添加到该溶液中并搅拌该溶液直至其完全溶解。
[0254]
在混合浆料的同时,在10分钟内添加2.4kg的磷酸溶液。在整个实验的过程中,悬浮液的温度保持在70℃
±
1℃。最后,在添加酸之后,将悬浮液再搅拌5分钟,然后将其从容器中取出并使其冷却。
[0255]
所获得的经表面反应碳酸钙的体积确定的中值粒子尺寸d
50
为4.0μm,体积确定的顶切粒子尺寸d
98
为9.1μm,并且比表面积ssa为93.1m2g
‑1。
[0256]
实施例7
[0257]
经表面反应碳酸钙通过如下方式获得:通过调节来自挪威hustadmarmor的研磨大理石碳酸钙的固含量以使得获得基于水性悬浮液的总重量计为20%重量的固含量,在混合容器中制备10升研磨碳酸钙的水性悬浮液。该研磨碳酸钙具有通过沉降测定的90%小于2μm的重量基粒子尺寸分布,d
50
(重量)为0.7μm且d
98
(重量)为3.4μm。
[0258]
另外,将磷酸稀释以使其包含基于溶液总重量计为30%的磷酸。然后将基于纯磷酸总重量计为13.8%重量的mgso4·
7h2o添加到该溶液中并搅拌该溶液直至其完全溶解。
[0259]
在混合浆料的同时,在10分钟内添加2.6kg的磷酸溶液。在整个实验的过程中,悬浮液的温度保持在70℃
±
1℃。最后,在添加酸之后,将悬浮液再搅拌5分钟,然后将其从容器中取出并使其冷却。
[0260]
所获得的经表面反应碳酸钙的体积确定的中值粒子尺寸d
50
为3.8μm,体积确定的顶切粒子尺寸d
98
为7.7μm,并且比表面积ssa为92.7m2g
‑1。
[0261]
实施例8
[0262]
经表面反应碳酸钙通过如下方式获得:通过调节来自挪威hustadmarmor的研磨大理石碳酸钙的固含量以使得获得基于水性悬浮液的总重量计为20%重量的固含量,在混合容器中制备10升研磨碳酸钙的水性悬浮液。该研磨碳酸钙具有通过沉降测定的90%小于2μm的重量基粒子尺寸分布,d
50
(重量)为0.7μm且d
98
(重量)为3.4μm。
[0263]
另外,将磷酸稀释以使其包含基于溶液总重量计为30%的磷酸。然后将基于纯磷酸总重量计为16.4%重量的mgbr2·
6h2o添加到该溶液中并搅拌该溶液直至其完全溶解。
[0264]
在混合浆料的同时,在10分钟内添加2.4kg的磷酸溶液。在整个实验的过程中,悬浮液的温度保持在70℃
±
1℃。最后,在添加酸之后,将悬浮液再搅拌5分钟,然后将其从容器中取出并使其冷却。
[0265]
所获得的经表面反应碳酸钙的体积确定的中值粒子尺寸d
50
为4.2μm,体积确定的顶切粒子尺寸d
98
为9.7μm,并且比表面积ssa为70.9m2g
‑1。
[0266]
实施例9
[0267]
经表面反应碳酸钙通过如下方式获得:通过调节来自挪威hustadmarmor的研磨大理石碳酸钙的固含量以使得获得基于水性悬浮液的总重量计为20%重量的固含量,在混合容器中制备10升研磨碳酸钙的水性悬浮液。该研磨碳酸钙具有通过沉降测定的90%小于2μm的重量基粒子尺寸分布,d
50
(重量)为0.7μm且d
98
(重量)为3.4μm。
[0268]
另外,将磷酸稀释以使其包含基于溶液总重量计为30%的磷酸。然后将基于纯磷酸总重量计为49.3%重量的mgbr2·
6h2o添加到该溶液中并搅拌该溶液直至其完全溶解。
[0269]
在混合浆料的同时,在10分钟内添加2.6kg的磷酸溶液。在整个实验的过程中,悬浮液的温度保持在70℃
±
1℃。最后,在添加酸之后,将悬浮液再搅拌5分钟,然后将其从容器中取出并使其冷却。
[0270]
所获得的经表面反应碳酸钙的体积确定的中值粒子尺寸d
50
为3.9μm,体积确定的顶切粒子尺寸d
98
为8.4μm,并且比表面积ssa为72.8m2g
‑1。
[0271]
结果
[0272]
如上所述,制备的经表面反应碳酸钙粒子在它们的粒子尺寸分布、它们的比表面积和它们的孔隙率方面进行表征。结果以及所采用的镁盐及其浓度汇总在下表1中。此外,对根据实施例1和3至5制备的经表面反应碳酸钙粒子进行xrd测量以确定它们的晶体结构(参见表2)。
[0273]
从表1中可以看出,与对比实施例1和5相比,水溶性无机镁盐的添加导致bet值的显著增加。从所述实验数据还可以看出,bet的增加可通过选择特定的镁盐来控制。
[0274]
此外,表1还表明,该经表面反应碳酸钙的粒子内侵入式比孔容能够以预定方式改变。尽管4.1mmol mg
2+
/g caco3的较低浓度导致粒子内侵入式比孔容增加,但与对比实施例1和5相比,8.2mmol mg
2+
/g caco3或更高的镁盐浓度的添加导致粒子内侵入式比孔容低于对比实施例的粒子内侵入式比孔容。还注意到,该经表面反应碳酸钙粒子的粒子尺寸分布没有受到显著影响,如从表1中的d
50
和d
98
值可以看出的。
[0275]
汇总在表2中的实施例1和3至5的经表面反应碳酸钙粒子的xrd分析揭示出:本发明的样品(实施例3和4)含有更高量的羟基磷灰石,这表明在本发明的粒子中已经形成更多的结晶磷酸钙,仅留下非常少的无定形磷酸钙。换言之,本发明的经表面反应碳酸钙粒子表现出更高的结晶度。此外,在本发明实施例3至4中获得的经表面反应碳酸钙中可清楚地检
测到镁矿物白磷钙石。
[0276]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1