异氰酸酯系粘接剂的制作方法
1.本发明涉及异氰酸酯系粘接剂。
背景技术:
2.以往,在汽车的车身、前门、后门、后背门、前保险杠、后保险杠、门槛板镶条等汽车的内外装构件中,使用了钢板,但从谋求汽车的轻量化的观点考虑,一部分使用聚丙烯树脂基材等结晶性热塑性树脂基材(以下,将“结晶性热塑性树脂基材”也简称为“树脂基材”)的情况增加。
3.这样在汽车的内外装构件中使用树脂的情况下,通常,进行对树脂基材的粘接面涂布底漆后涂布粘接剂进行贴合的操作。此外,为了省去底漆的涂布,也提出了对树脂基材的表面实施表面处理后,涂布粘接剂进行贴合的方法(例如,专利文献1)。
4.现有技术文献
5.专利文献
6.专利文献1:日本特开2014-25000号公报
技术实现要素:
7.发明所要解决的课题
8.在这种情况下,本发明人等以专利文献1作为参考,对实施了各种表面处理的树脂基材使用各种异氰酸酯系粘接剂,结果明确了异氰酸酯系粘接剂的粘接性(初始粘接性)、耐水粘接性、或耐热粘接性不一定为可以令人满意的水平。
9.因此,本发明鉴于上述情况,其目的是提供显示优异的粘接性、耐水粘接性和耐热粘接性的异氰酸酯系粘接剂。
10.用于解决课题的方法
11.本发明人等对上述课题进行了深入研究,结果认识到,将异氰酸酯系粘接剂在特定的条件下进行了固化时的物性、与树脂基材的表面状态的平衡是重要的,从而完成了本发明。
12.即,本发明人等发现,通过以下构成可以解决上述课题。
13.(1)一种异氰酸酯系粘接剂,是实施了表面处理的结晶性热塑性树脂基材所使用的异氰酸酯系粘接剂,
14.上述异氰酸酯系粘接剂通过在23℃、50%rh的条件下静置3天而固化后,后述式(a)所示的值为2.0~70,
15.上述结晶性热塑性树脂基材的后述式(b)所示的值为2.0~30.0。
16.(2)根据上述(1)所述的异氰酸酯系粘接剂,上述异氰酸酯系粘接剂具有主剂和固化剂,所述主剂含有氨基甲酸酯预聚物,所述固化剂含有重均分子量为1000以下的3官能以上的多元醇,
17.上述固化剂的混合量相对于上述主剂的混合量的比例以质量比计为1.1以下。
18.(3)根据上述(1)或(2)所述的异氰酸酯系粘接剂,上述异氰酸酯系粘接剂含有选自脂肪族异氰酸酯的异氰脲酸酯体、脂肪族异氰酸酯的脲基甲酸酯体、和脂肪族异氰酸酯的缩二脲体中的至少1种脂肪族异氰酸酯改性体。
19.(4)根据上述(1)~(3)中任一项所述的异氰酸酯系粘接剂,上述异氰酸酯系粘接剂含有硅烷偶联剂。
20.(5)根据上述(1)~(4)中任一项所述的异氰酸酯系粘接剂,上述异氰酸酯系粘接剂含有炭黑或碳酸钙。
21.发明的效果
22.如以下所示那样,根据本发明,可以提供显示优异的粘接性、耐水粘接性和耐热粘接性的异氰酸酯系粘接剂。
具体实施方式
23.以下,对本发明的异氰酸酯系粘接剂进行说明。
24.另外,在本说明书中,使用“~”表示的数值范围是指包含“~”的前后所记载的数值作为下限值和上限值的范围。
25.此外,各成分可以单独使用1种,也可以并用2种以上。这里,关于各成分,在并用2种以上的情况下,关于该成分,所谓含量,只要没有特别指明,就是指合计含量。
26.此外,在本发明的异氰酸酯系粘接剂为具有主剂和固化剂的双组分型异氰酸酯系粘接剂的情况下,本发明的异氰酸酯系粘接剂中的各成分的所谓含量,是指相对于主剂和固化剂的合计的含量。
27.本发明的异氰酸酯系粘接剂是下述异氰酸酯系粘接剂,其用于实施了表面处理的结晶性热塑性树脂基材,上述异氰酸酯系粘接剂通过在23℃、50%rh(相对湿度)的条件下静置3天而固化后,后述式(a)所示的值为2.0~70,上述结晶性热塑性树脂基材的后述式(b)所示的值为2.0~30.0。
28.[异氰酸酯系粘接剂]
[0029]
异氰酸酯系粘接剂只要是通过在23℃、50%rh的条件下静置3天而固化后,后述式(a)所示的值为2.0~70的异氰酸酯系粘接剂(以下,也称为“本发明的粘接剂”),就没有特别限制。
[0030]
基于粘接性、粘接耐久性(例如,70℃加热10分钟后的强度)、耐水粘接性和耐热粘接性更优异的理由,上述异氰酸酯系粘接剂优选为单组分型异氰酸酯系粘接剂或双组分型异氰酸酯系粘接剂,更优选为双组分型异氰酸酯系粘接剂。以下,将“粘接性、粘接耐久性(例如,70℃加热10分钟后的强度)、耐水粘接性和耐热粘接性更优异”也称为“本发明的效果等更优异”。
[0031]
基于本发明的效果等更优异的理由,上述异氰酸酯系粘接剂优选为氨基甲酸酯系粘接剂。
[0032]
这里,所谓异氰酸酯系粘接剂,是指包含异氰酸酯(具有异氰酸酯基的化合物)的粘接剂。此外,所谓氨基甲酸酯系粘接剂,是指包含氨基甲酸酯(具有氨基甲酸酯键的化合物)的粘接剂。
[0033]
作为单组分型异氰酸酯系粘接剂,可举出例如包含氨基甲酸酯预聚物的湿固化型
粘接剂。
[0034]
作为双组分型异氰酸酯系粘接剂,可举出例如,含有包含氨基甲酸酯预聚物的主剂、和包含多元醇的固化剂的粘接剂。
[0035]
〔氨基甲酸酯预聚物〕
[0036]
可举出氨基甲酸酯预聚物为1分子内在分子末端含有多个异氰酸酯基的氨基甲酸酯预聚物作为优选的方案之一。
[0037]
作为氨基甲酸酯预聚物,可以使用以往公知的物质。例如,可以使用通过使多异氰酸酯与1分子中具有2个以上含活性氢基的化合物(以下,也称为“活性氢化合物”。)以异氰酸酯基相对于含活性氢基变得过剩的方式反应而获得的反应生成物等。
[0038]
在本发明中,含活性氢基是指含有活性氢的基团。作为含活性氢基,可举出例如,羟基、氨基、亚氨基。
[0039]
<多异氰酸酯>
[0040]
制造氨基甲酸酯预聚物时所使用的多异氰酸酯只要是分子内具有2个以上异氰酸酯基的物质,就没有特别限定。
[0041]
作为多异氰酸酯,可举出例如,甲苯二异氰酸酯(tdi。例如,2,4-甲苯二异氰酸酯、2,6-甲苯二异氰酸酯)、二苯基甲烷二异氰酸酯(mdi。例如,4,4
’‑
二苯基甲烷二异氰酸酯、2,4
’‑
二苯基甲烷二异氰酸酯)、1,4-苯二异氰酸酯、多亚甲基多亚苯基多异氰酸酯、苯二亚甲基二异氰酸酯(xdi)、四甲基苯二亚甲基二异氰酸酯(tmxdi)、联甲苯胺二异氰酸酯(todi)、1,5-萘二异氰酸酯(ndi)、三苯基甲烷三异氰酸酯那样的芳香族多异氰酸酯;1,6-己二异氰酸酯(hdi)、三甲基六亚甲基二异氰酸酯(tmhdi)、赖氨酸二异氰酸酯、降冰片烷二异氰酸酯(nbdi)、反式环己烷-1,4-二异氰酸酯、异佛尔酮二异氰酸酯(ipdi)、双(异氰酸酯基甲基)环己烷(h6xdi)、二环己基甲烷二异氰酸酯(h
12
mdi)那样的、脂肪族和/或脂环式的多异氰酸酯;它们的碳二亚胺改性多异氰酸酯、异氰脲酸酯改性多异氰酸酯、脲基甲酸酯改性体。
[0042]
基于固化性优异的理由,多异氰酸酯优选为芳香族多异氰酸酯,更优选为mdi。
[0043]
多异氰酸酯可以分别单独使用或组合使用2种以上。
[0044]
<活性氢化合物>
[0045]
制造氨基甲酸酯预聚物时所使用的1分子中具有2个以上含活性氢基的化合物(活性氢化合物)没有特别限定。作为含活性氢基,可举出例如,羟(oh)基、氨基、亚氨基。
[0046]
作为上述活性氢化合物,适合举出例如,1分子中具有2个以上羟(oh)基的多元醇、1分子中具有2个以上氨基和/或亚氨基的多胺化合物等,其中,优选为多元醇。
[0047]
活性氢化合物可以分别单独使用或组合使用2种以上。
[0048]
<含量>
[0049]
在本发明的粘接剂中,基于本发明的效果等更优异的理由,本发明的粘接剂中,氨基甲酸酯预聚物的含量优选为10~95质量%,更优选为20~60质量%。
[0050]
〔多元醇〕
[0051]
多元醇只要是具有2个以上oh基的(即,2官能以上的)化合物,就没有特别限定。作为多元醇的具体例,可举出聚醚多元醇;聚酯多元醇;丙烯酸系多元醇;聚丁二烯多元醇、被加氢了的聚丁二烯多元醇;低分子多元醇类;它们的混合多元醇。其中,基于本发明的效果
等更优异的理由,优选为聚醚多元醇。
[0052]
作为上述聚醚多元醇的具体例,可举出聚氧乙烯二醇(聚乙二醇)、聚氧丙烯二醇(聚丙二醇:ppg)、聚氧丙烯三醇、氧化乙烯/氧化丙烯共聚物、聚四亚甲基醚二醇(ptmeg)、聚四甘醇等。其中,基于本发明的效果等更优异的理由,优选为聚氧丙烯二醇(聚丙二醇:ppg)、聚氧丙烯三醇、氧化乙烯/氧化丙烯共聚物,聚氧丙烯三醇。
[0053]
基于本发明的效果等更优异的理由,多元醇优选为3官能的多元醇(即,具有3个以上oh基的多元醇),更优选为聚氧丙烯三醇。
[0054]
基于本发明的效果等更优异的理由,多元醇的重均分子量(mw)优选为1000以下。上述mw的下限没有特别限制,但基于本发明的效果等更优异的理由,优选为100以上。
[0055]
另外,在本说明书中,重均分子量为通过gpc法(溶剂:四氢呋喃(thf))而获得的聚苯乙烯换算值。
[0056]
基于本发明的效果等更优异的理由,多元醇优选为重均分子量1000以下的3官能以上的多元醇(以下,也称为“特定多元醇”)。
[0057]
<含量>
[0058]
在本发明的粘接剂中,基于本发明的效果等更优异的理由,本发明的粘接剂中,多元醇的含量优选为1~90质量%,更优选为5~50质量%。
[0059]
〔混合比〕
[0060]
在本发明的粘接剂为具有含有氨基甲酸酯预聚物的主剂和含有多元醇的固化剂的双组分型异氰酸酯系粘接剂的情况下,基于本发明的效果等更优异的理由,固化剂的混合量相对于主剂的混合量的比例(固化剂/主剂)以质量比计优选为1.1以下。上述比例(固化剂/主剂)的下限没有特别限制,但基于本发明的效果等更优异的理由,优选为0.01以上,更优选为0.05以上。
[0061]
此外,在本发明的粘接剂为具有含有氨基甲酸酯预聚物的主剂和含有多元醇的固化剂的双组分型异氰酸酯系粘接剂的情况下,基于本发明的效果等更优异的理由,氨基甲酸酯预聚物所具有的异氰酸酯基(nco基)与多元醇所具有的oh基的摩尔比(nco/oh)优选为0.2~10.0,更优选为1.0~2.5。
[0062]
在本发明的粘接剂中,多元醇的含量没有特别限制,但基于本发明的效果等更优异的理由,相对于氨基甲酸酯预聚物的含量,优选为5~120质量%,更优选为10~100质量%。
[0063]
〔其它成分〕
[0064]
本发明的粘接剂可以含有除上述成分以外的其它成分。
[0065]
在双组分型的情况下,可以适当选择将其它成分添加在主剂和固化剂的任一者中。
[0066]
作为那样的其它成分,可以进一步含有例如,填充剂(例如,炭黑、碳酸钙)、脂肪族异氰酸酯的改性体(例如,异氰脲酸酯体、脲基甲酸酯体、缩二脲体)、催化剂(固化催化剂)、增塑剂、防老剂、抗氧化剂、硅烷偶联剂、颜料(染料)、增粘剂、松油醇那样的萜化合物、触变性赋予剂、紫外线吸收剂、阻燃剂、表面活性剂(包含流平剂)、分散剂、脱水剂、抗静电剂等各种添加剂等。
[0067]
另外,上述填充剂例如可以利用选自脂肪酸、树脂酸、氨基甲酸酯化合物和脂肪酸
酯中的至少1种处理剂进行表面处理。
[0068]
此外,在本发明的粘接剂为双组分型的情况下,可以适当选择将上述任意成分添加在主剂和固化剂的任一者中。
[0069]
基于本发明的效果等更优异的理由,本发明的粘接剂优选不含有萜化合物。
[0070]
<脂肪族异氰酸酯的改性体>
[0071]
基于本发明的效果等更优异的理由,本发明的粘接剂优选含有选自脂肪族异氰酸酯的异氰脲酸酯体(异氰脲酸酯体(
ヌレート
体))、脂肪族异氰酸酯的脲基甲酸酯体、和脂肪族异氰酸酯的缩二脲体中的至少1种脂肪族异氰酸酯改性体。
[0072]
在本发明的粘接剂中,脂肪族异氰酸酯的改性体的含量没有特别限制,但基于本发明的效果等更优异的理由,本发明的粘接剂中,优选为0.1~10质量%,更优选为0.5~5质量%。
[0073]
在本发明的粘接剂中,脂肪族异氰酸酯的改性体的含量没有特别限制,但基于本发明的效果等更优异的理由,相对于氨基甲酸酯预聚物的含量,优选为1~20质量%,更优选为2~10质量%。
[0074]
<硅烷偶联剂>
[0075]
基于本发明的效果等更优异的理由,本发明的粘接剂优选含有硅烷偶联剂。
[0076]
在本发明的粘接剂中,硅烷偶联剂的含量没有特别限制,但基于本发明的效果等更优异的理由,相对于氨基甲酸酯预聚物的含量,优选为0.01~10质量%,更优选为0.1~5质量%。
[0077]
<炭黑>
[0078]
基于本发明的效果等更优异的理由,本发明的粘接剂优选含有炭黑。
[0079]
炭黑没有特别限制。可举出例如,saf(超耐磨炉黑,super abrasion furnace)、isaf(中超耐磨炉黑,intermediate super abrasion furnace)、haf(高耐磨炉黑,high abrasion furnace)、fef(快压出炉黑,fast extruding furnace)、gpf(通用炉黑,general purpose furnace)、srf(半补强炉黑,semi-reinforcing furnace)、ft(细热裂黑,fine thermal)、mt(中等热裂黑,medium thermal)等。
[0080]
具体而言,作为上述saf,可例示
シースト
9(東海
カーボン
社制),作为isaf,可例示
ショウワブラック
n220(昭和
キャボット
社制),作为haf,可例示
シースト
3(東海
カーボン
社制)、
ニテロン
#200(新日化
カーボン
社制),作为fef,可例示htc#100(中部
カーボン
社制)等。此外,作为gpf,可例示旭#55(旭
カーボン
社制)、
シースト
5(東海
カーボン
社制),作为srf,可例示旭#50(旭
カーボン
社制)、三菱#5(三菱化学社制),作为ft,可例示旭
サーマル
(旭
カーボン
社制)、htc#20(中部
カーボン
社制),作为mt,可例示旭#15(旭
カーボン
社制)等。
[0081]
在本发明的粘接剂中,炭黑的含量没有特别限制,但基于本发明的效果等更优异的理由,相对于氨基甲酸酯预聚物的含量,优选为30~70质量%,更优选为40~60质量%。
[0082]
<碳酸钙>
[0083]
基于本发明的效果等更优异的理由,本发明的粘接剂优选含有碳酸钙。
[0084]
碳酸钙没有特别限制。可举出例如,重质碳酸钙、沉降性碳酸钙(轻质碳酸钙)、胶态碳酸钙等。
[0085]
在本发明的粘接剂中,碳酸钙的含量没有特别限制,但基于本发明的效果等更优异的理由,相对于氨基甲酸酯预聚物的含量,优选为20~150质量%,更优选为20~120质量%,进一步优选为30~70质量%。
[0086]
<催化剂(固化催化剂)>
[0087]
基于本发明的效果等更优异的理由,本发明的粘接剂优选含有固化催化剂。
[0088]
固化催化剂没有特别限定,作为具体例,可举出2-乙基己酸、油酸等羧酸类;多磷酸、酸式磷酸乙酯、酸式磷酸丁酯等磷酸类;辛酸铋等铋催化剂;二丁基二月桂酸锡、二辛基二月桂酸锡等锡催化剂;1,4-二氮杂二环[2.2.2]辛烷、2,4,6-三(二甲基氨基甲基)苯酚(例如,dmp-30)、包含二吗啉代二乙基醚结构的化合物等叔胺催化剂等。
[0089]
在粘接性更优异这方面,固化催化剂优选包含二吗啉代二乙基醚结构。
[0090]
二吗啉代二乙基醚结构为以二吗啉代二乙基醚作为基本骨架的结构。
[0091]
在二吗啉代二乙基醚结构中,吗啉环所具有的氢原子可以被取代基取代。取代基没有特别限制。可举出例如,烷基。作为烷基,可举出例如,甲基、乙基。
[0092]
固化催化剂可以分别单独使用或组合使用2种以上。
[0093]
在本发明的粘接剂中,固化催化剂的含量没有特别限制,但基于本发明的效果等更优异的理由,相对于氨基甲酸酯预聚物的含量,优选为0.05~2.0质量%,更优选为0.1~0.5质量%。
[0094]
<增塑剂>
[0095]
基于本发明的效果等更优异的理由,本发明的粘接剂优选含有增塑剂。
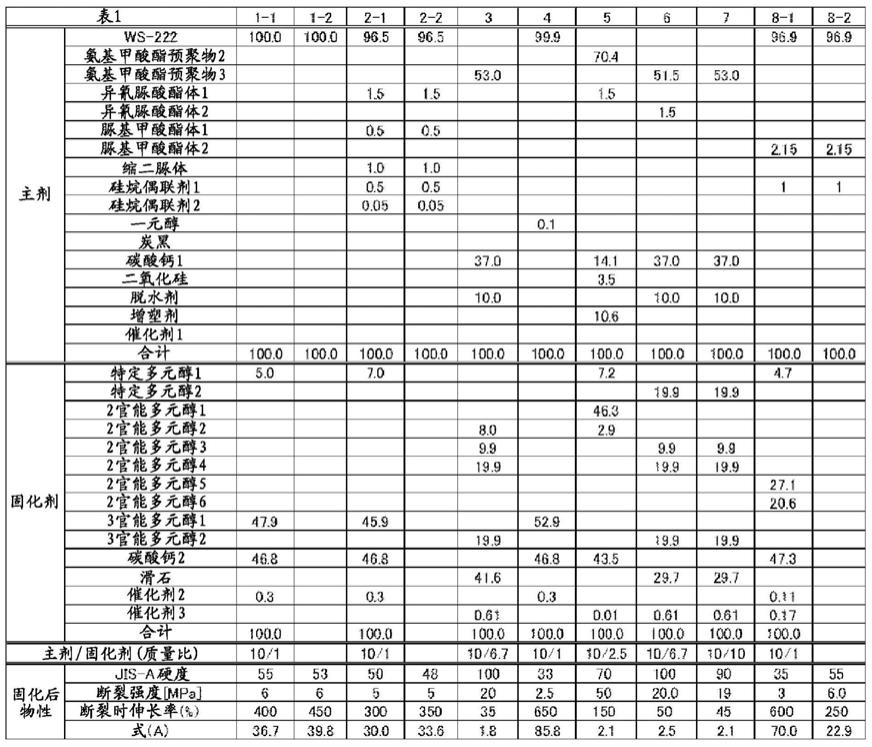
[0096]
作为增塑剂的具体例,可举出邻苯二甲酸二异壬酯(dinp);己二酸二辛酯、琥珀酸异癸酯;二甘醇二苯甲酸酯、季戊四醇酯;油酸丁酯、乙酰蓖麻油酸甲酯;磷酸三甲苯酯、磷酸三辛酯;己二酸丙二醇聚酯、己二酸丁二醇聚酯等,它们可以单独使用1种,也可以并用2种以上。
[0097]
在本发明的粘接剂中,增塑剂的含量没有特别限制,但基于本发明的效果等更优异的理由,相对于氨基甲酸酯预聚物的含量,优选为1~50质量%,更优选为5~40质量%。
[0098]
〔式(a)〕
[0099]
如上所述,本发明的粘接剂通过在23℃、50%rh的环境下静置3天而固化(以下,也称为“特定固化”)后,下述式(a)所示的值为2.0~70。以下,将“式(a)所示的值”也简称为“式(a)”。
[0100]
式(a)=(jis-a硬度)/(断裂强度[mpa])
×
(断裂时伸长率(%))/100
[0101]
式(a)是关于固化后的粘接剂,将jis-a硬度除以断裂强度[mpa],对所得的值乘以断裂时伸长率(%),将所得的值除以100而得的值。
[0102]
<jis-a硬度>
[0103]
所谓jis-a硬度,是按照jis k6253所规定的硬度计硬度试验,在温度20℃、55%rh的环境下使用a型的硬度计测定的硬度。
[0104]
基于本发明的效果等更优异的理由,jis-a硬度优选为10~90,更优选为20~85,进一步优选为30~80,特别优选为40~70。
[0105]
<断裂强度>
[0106]
所谓断裂强度,是在按照jis k6850:1999进行了拉伸试验(拉伸速度50mm/分钟,
20℃、50%rh的环境下)时的断裂强度。
[0107]
基于本发明的效果等更优异的理由,断裂强度优选为0.1~100mpa,更优选为1~50mpa。
[0108]
<断裂时伸长率>
[0109]
所谓断裂时伸长率,是在按照jis k6850:1999进行了拉伸试验(拉伸速度50mm/分钟,20℃、50%rh的环境下)时的断裂时伸长率。
[0110]
基于本发明的效果等更优异的理由,断裂时伸长率优选为10~1000%,更优选为50~800%,进一步优选为100~500%。
[0111]
<适合的方案>
[0112]
基于本发明的效果等更优异的理由,式(a)优选为5~60,更优选为10~50,进一步优选为20~40。
[0113]
作为获得在特定固化后式(a)为2.0~70的异氰酸酯系粘接剂的方法,可举出例如,关于变更了成分的种类、量的各种异氰酸酯系粘接剂,评价特定固化后的jis-a硬度、断裂强度和断裂时伸长率,求出式(a),获得式(a)落入2.0~70的范围的异氰酸酯系粘接剂的方法等。
[0114]
[结晶性热塑性树脂基材]
[0115]
如上所述,上述本发明的粘接剂用于实施了表面处理的结晶性热塑性树脂基材(树脂基材)。这里,上述树脂基材的后述式(b)所示的值为2.0~30.0。
[0116]
以下,将实施了表面处理、并且后述式(b)所示的值为1.0~10的树脂基材也称为“本发明的树脂基材”。
[0117]
〔材料〕
[0118]
作为本发明的树脂基材的材料的具体例,可举出聚乙烯、聚丙烯、聚丁烯等聚烯烃系树脂;聚甲基丙烯酸甲酯等甲基丙烯酸系树脂;聚苯乙烯、abs、as等聚苯乙烯系树脂;聚对苯二甲酸乙二醇酯(pet)、聚对苯二甲酸丁二醇酯(pbt)、聚对苯二甲酸丙二醇酯、聚萘二甲酸乙二醇酯(pen)、聚对苯二甲酸1,4-环己基二甲醇酯(pct)等聚酯系树脂;选自聚己酰胺(尼龙6)、聚己二酰己二胺(尼龙66)、聚癸二酰己二胺(尼龙610)、聚十二烷二酰己二胺(尼龙612)、聚十二烷酰胺(尼龙12)、聚对苯二甲酰己二胺(尼龙6t)、聚间苯二甲酰己二胺(尼龙6i)、聚己酰胺/聚对苯二甲酰己二胺共聚物(尼龙6/6t)、聚己二酰己二胺/聚对苯二甲酰己二胺共聚物(尼龙66/6t)、聚己二酰己二胺/聚间苯二甲酰己二胺共聚物(尼龙66/6i)等尼龙树脂和尼龙共聚物树脂中的聚酰胺树脂;聚氯乙烯树脂;聚甲醛(pom);聚碳酸酯(pc)树脂;聚苯硫醚(pps)树脂;改性聚苯醚(ppe)树脂;聚醚酰亚胺(pei)树脂;聚砜(psf)树脂;聚醚砜(pes)树脂;聚酮树脂;聚醚腈(pen)树脂;聚醚酮(pek)树脂;聚醚醚酮(peek)树脂;聚醚酮酮(pekk)树脂;聚酰亚胺(pi)树脂;聚酰胺酰亚胺(pai)树脂;氟树脂;使这些树脂改性而得的改性树脂或这些树脂的混合物等。其中,基于本发明的效果等更优异的理由,优选为聚烯烃系树脂,更优选为聚乙烯、聚丙烯,进一步优选为聚丙烯。本发明的树脂基材可以含有2种以上树脂。
[0119]
〔表面处理〕
[0120]
在本发明的树脂基材中,关于对树脂基材实施的表面处理,只要后述式(b)所示的值为2.0~30.0的范围内,就没有特别限制,但基于本发明的效果等更优异的理由,优选为
选自电晕处理、等离子体处理、火焰处理、itro处理、uv处理(紫外线照射处理)、和受激准分子处理中的至少1种,更优选为火焰处理、等离子体处理、电晕处理、itro处理,进一步优选为火焰处理、等离子体处理。
[0121]
<火焰处理>
[0122]
火焰处理是通过火焰(焰)进行表面处理的方法。
[0123]
火焰处理可以使用:利用燃烧器的方法等以往公知的方法。
[0124]
火焰处理的气体压力优选为0.005~10mpa,更优选为0.01~1.5mpa。
[0125]
火焰处理的速度优选为100~2000mm/秒,更优选为200~1000mm/秒。
[0126]
在使用燃烧器进行火焰处理的情况下,燃烧器与树脂基材的表面的距离优选为10~600mm,更优选为20~400mm。
[0127]
<等离子体处理>
[0128]
等离子体处理是通过等离子体放电进行表面处理的方法。
[0129]
等离子体处理没有特别限制,可举出例如,大气压等离子体处理、真空等离子体处理等。
[0130]
等离子体处理所使用的等离子体气体(工艺气体)没有特别限制,可例示氮气、氦气、氩气等、在这些气体中混合了氧气、二氧化碳气体和氢气中的1种以上而得的混合气体等。
[0131]
等离子体处理的速度优选为10~1500mm/秒,更优选为50~1000mm/秒。
[0132]
在使用等离子体放电喷嘴进行等离子体处理的情况下,等离子体放电喷嘴与树脂基材的表面的距离优选为1~100mm,更优选为5~50mm。
[0133]
<uv处理>
[0134]
uv处理是通过紫外线(uv)照射进行表面处理的方法。
[0135]
基于本发明的效果等更优异的理由,照射uv的时间优选为20秒以上,更优选为60秒以上,进一步优选为90秒以上,特别优选为120秒以上。基于本发明的效果等更优异的理由,上限优选为900秒以下,更优选为300秒以下。
[0136]
在使用uv照射装置进行uv处理的情况下,uv处理装置与树脂基材的表面的距离优选为1~100mm,更优选为5~50mm。
[0137]
<电晕处理>
[0138]
电晕处理是通过电晕放电进行表面处理的方法。
[0139]
基于本发明的效果等更优异的理由,电晕处理的速度优选为10~1000mm/秒,更优选为20~500mm/秒。
[0140]
在使用电晕放电喷嘴进行电晕处理的情况下,基于本发明的效果等更优异的理由,电晕放电喷嘴与树脂基材的表面的距离优选为1~100mm,更优选为5~50mm。
[0141]
<itro处理>
[0142]
itro处理是通过向用于形成火焰(焰)的燃料气体中导入硅烷化合物等,使用该火焰对表面实施处理从而在表面形成纳米级的氧化硅膜,使表面与粘接剂的密合性提高的处理。
[0143]
基于本发明的效果等更优异的理由,itro处理的气体压力优选为0.005~10mpa,基于本发明的效果等更优异的理由,更优选为0.01~1.5mpa。
[0144]
基于本发明的效果等更优异的理由,itro处理的速度优选为100~2000mm/秒,更优选为200~1000mm/秒。
[0145]
在使用燃烧器进行itro处理的情况下,基于本发明的效果等更优异的理由,燃烧器与树脂基材的表面的距离优选为1~600mm,更优选为20~400mm。
[0146]
〔式(b)〕
[0147]
如上所述,本发明的树脂基材的下述式(b)所示的值为2.0~30.0。以下,将“式(b)所示的值”也简称为“式(b)”。
[0148]
式(b)=δd/δ
p
+δ
p
[0149]
其中,δ
p
=γ
p-γ
p0
,δd=|γ
d-γ
d0
|,
[0150]
γ
p0
为表面处理前的结晶性热塑性树脂基材的表面自由能的极性项,
[0151]
γ
p
为表面处理后的结晶性热塑性树脂基材的表面自由能的极性项,
[0152]
γ
d0
为表面处理前的结晶性热塑性树脂基材的表面自由能的分散项,
[0153]
γd为表面处理后的结晶性热塑性树脂基材的表面自由能的分散项。
[0154]
式(b)为δd除以δ
p
而得的值与δ
p
之和。
[0155]
此外,δ
p
为从γ
p
减去γ
p0
而得的值。
[0156]
此外,δd为γd与γ
d0
之差的绝对值。
[0157]
基于本发明的效果等更优异的理由,δ
p
优选为1~50,更优选为5~20。
[0158]
基于本发明的效果等更优异的理由,δd优选为1~20,更优选为2~10。
[0159]
<适合的方案(其1)>
[0160]
基于本发明的效果等更优异的理由,式(b)优选为5.0~20.0,更优选为10.0~15.0。
[0161]
<适合的方案(其2)>
[0162]
在表面处理时的表面温度为大于或等于比树脂基材(表面处理前的树脂基材)的通过差示扫描量热(dsc)测定而获得的吸热峰的起点温度低50℃的温度的情况下,基于本发明的效果等更优异的理由,式(b)优选为5.0~30.0,更优选为10.0~25.0,进一步优选为10.0~15.0。
[0163]
此外,在表面处理时的表面温度为小于比树脂基材(表面处理前的树脂基材)的通过差示扫描量热(dsc)测定而获得的吸热峰的起点温度低50℃的温度的情况下,基于本发明的效果等更优异的理由,式(b)优选为2.0~5.0。
[0164]
另外,例如,如果是在后述的实施例中使用的树脂基材1,则通过dsc而获得的吸热峰的起点温度为120℃,因此比上述通过dsc而获得的吸热峰的起点温度低50℃的温度为70℃(=120℃-50℃)。
[0165]
此外,表面处理时的表面温度可以通过粘贴型的热电偶、通过热而变色的墨液和封条以及热成像等来测定。
[0166]
<表面自由能的求法>
[0167]
接下来,对表面自由能的求法进行说明。
[0168]
树脂基材的表面自由能的极性项和分散项可以按照owens and wendt法(j.appl.polym.sci.,13,1741-1747(1969).)而求出。
[0169]
即,在树脂基材表面滴加表面自由能已知的液体(试验液体),测定接触角,由基于
owens and wendt解析的计算式(下述式(1)~(3)),算出树脂基材的表面自由能的极性项和分散项。
[0170]
式(1):1+cosθ=2[(γd·
γ
ld
)/γ
l2
]
1/2
+2[(γ
p
·
γ
lp
)/γ
l2
]
1/2
[0171]
式(2):γ=γd+γ
p
[0172]
式(3):γ
l
=γ
ld
+γ
lp
[0173]
其中,各符号表示以下内容。
[0174]
·
θ:各试验液体的接触角
[0175]
·
γd:树脂基材的表面自由能的分散项[mjm-2
]
[0176]
·
γ
ld
:试验液体的表面自由能的分散项[mjm-2
]
[0177]
·
γ
p
:树脂基材的表面自由能的极性项[mjm-2
]
[0178]
·
γ
lp
:试验液体的表面自由能的极性项[mjm-2
]
[0179]
在本发明中,使用水和二碘甲烷(二碘甲烷)作为试验液体。作为试验液体而使用的水和二碘甲烷的表面自由能使用:水:γ
ld
=21.8mjm-2
,γ
lp
=51.0mjm-2
,二碘甲烷:γ
ld
=49.5mjm-2
,γ
lp
=1.3mjm-2
。通过将这些数值导入到上述式(1)~(3),将使用了各试验液体的接触角的数值导入到上述式,从而可以算出树脂基材的表面自由能的极性项和分散项。
[0180]
此外,上述接触角的测定如下:在25℃下,将试验液体滴加在树脂基材表面上,将从着滴到测定的等待时间设为5秒,通过按照在jis r3257中有记载的“静滴法”的θ/2法进行。作为接触角的测定装置,可以使用便携式接触角计(kruss社制)。
[0181]
关于表面处理前的树脂基材和表面处理后的树脂基材各自,通过如上所述算出表面自由能的极性项和分散项,可以求出上述式(b)。
[0182]
作为获得式(b)为2.0~30.0的实施了表面处理的树脂基材的方法,可举出例如,关于变更了树脂基材的材料、以及表面处理的种类和条件的实施了各种表面处理的树脂基材,算出表面处理前后的表面自由能的极性项和分散项,求出式(b),获得式(b)落入2.0~30.0的范围的实施了表面处理的树脂基材的方法等。
[0183]
[用途]
[0184]
本发明的异氰酸酯系粘接剂由于粘接性、耐水粘接性和耐热粘接性优异,因此对汽车的车身、前门、后门、后背门、前保险杠、后保险杠、门槛板镶条等汽车的内外装构件是特别有用的。
[0185]
[叠层体的制造方法]
[0186]
本发明的叠层体的制造方法(以下,也称为“本发明的制造方法”)是依次具备下述(1)~(3)的工序的叠层体的制造方法。另外,本发明的制造方法可以具备除下述(1)~(3)以外的工序。
[0187]
(1)表面处理工序
[0188]
对树脂基材的表面实施表面处理的工序
[0189]
(2)固化前粘接剂层形成工序
[0190]
通过向上述实施了表面处理的树脂基材上施与上述本发明的粘接剂,从而获得具有树脂基材和固化前粘接剂层的叠层体前体的工序
[0191]
(3)固化工序
[0192]
通过将上述固化前粘接剂层固化,从而获得具有树脂基材和固化后粘接剂层(以下,将“固化后粘接剂层”也简称为“粘接剂层”)的叠层体的工序
[0193]
以下,对各工序进行说明。
[0194]
〔表面处理工序〕
[0195]
表面处理工序为对树脂基材的表面实施表面处理的工序。
[0196]
实施了表面处理的树脂基材为上述本发明的树脂基材。即,实施了表面处理的树脂基材的上述式(b)所示的值为2.0~30.0。
[0197]
树脂基材的材料的具体例和适合的方案如上所述。
[0198]
表面处理的具体例和适合的方案如上所述。
[0199]
〔固化前粘接剂层形成工序〕
[0200]
固化前粘接剂层形成工序是通过在上述表面处理工序中获得的实施了表面处理的树脂基材(本发明的树脂基材)上施与上述本发明的粘接剂,从而获得具有树脂基材和固化前粘接剂层的叠层体前体的工序。也可以在本发明的树脂基材上涂布底漆后施与本发明的粘接剂。
[0201]
在实施了表面处理的树脂基材上施与本发明的粘接剂的方法没有特别限定,可举出例如,浸渍涂布法、采用双辊涂布机、狭缝涂布机、气刀涂布机、线棒涂布机、滑动料斗、喷射涂布、刮板涂布机、刮刀涂布机、挤压涂布机、逆转辊涂布机、转送辊涂布机、挤出涂布机、帘涂机、浸渍涂布机、模涂机、凹版辊进行的涂覆法、丝网印刷法、浸渍涂布法、喷涂法、旋转涂布法、喷墨法等。
[0202]
所形成的固化前粘接剂层的厚度没有特别限制,但优选为0.1~20mm。
[0203]
〔贴合工序〕
[0204]
本发明的制造方法在上述固化前粘接剂层形成工序与后述固化工序之间,可以进一步具备在上述固化前粘接剂层形成工序中形成的固化前粘接剂层的另一个面贴合其它基材(优选为上述实施了表面处理的同种的另外的树脂基材)(可以涂布底漆),从而获得依次具备树脂基材、固化前粘接剂层和树脂基材的叠层体前体的工序。
[0205]
〔固化工序〕
[0206]
固化工序是通过将在上述固化前粘接剂层形成工序中形成的固化前粘接剂层固化,从而获得具有树脂基材和固化后粘接剂层(粘接剂层)的叠层体(在具备上述贴合工序的情况下,为具有树脂基材、粘接剂层和树脂基材的叠层体)的工序。
[0207]
将固化前粘接剂层固化的方法没有特别限制,可举出在室温下放置的方法、进行加热的方法等。
[0208]
将固化前粘接剂层固化的方法不限于上述式(a)中的固化(即,在23℃、50%rh的条件下静置3天)。
[0209]
实施例
[0210]
以下,通过实施例对本发明进一步详细地说明,但本发明不限定于此。
[0211]
〔异氰酸酯系粘接剂的调制〕
[0212]
将下述表1的主剂栏所示的各成分以该表所示的比例(质量份)混合。
[0213]
此外,将该表的固化剂栏所示的各成分以该表所示的比例(质量份)进行了混合。这样操作而调制出1-1、1-2、2-1、2-2、3、4、5、6、7、8-1、8-2的粘接剂。
[0214]
另外,具有主剂和固化剂两者的是双组分型,在固化后形成氨基甲酸酯键。此外,仅具有主剂的是单组分型,在固化后形成脲键。
[0215]
例如,1-1由于具有主剂和固化剂两者,因此是双组分型,1-2由于仅具有主剂,因此是单组分型。
[0216]
在表1中显示各粘接剂的固化后(在23℃、50%rh的条件下静置3天)的“jis-a硬度”、“断裂强度[mpa]”和“断裂时伸长率(%)”。测定方法如上所述。另外,在粘接剂为双组分型的情况下,以表1的“主剂/固化剂(质量比)”栏所记载的比例混合。
[0217]
此外,在表1中显示各粘接剂的式(a)。
[0218]
表1
[0219][0220]
表1中的各成分的详细内容如下所述。
[0221]
·
ws-222:包含氨基甲酸酯预聚物的湿固化型粘接剂(横浜
ゴム
社制)
[0222]
·
氨基甲酸酯预聚物2:将聚丁二醇(三菱
ケミカル
社制ptmg650,重均分子量650)与聚氧丙烯二醇(三洋化成工业社制
サンニックス
pp2000,重均分子量2,000)与mdi(二苯基甲烷二异氰酸酯)(住化
バイエルウレタン
社制
スミジュール
44s)以nco/oh(摩尔比)成为2.0的方式混合,使混合物在80℃的条件下反应5小时而制造的氨基甲酸酯预聚物
[0223]
·
氨基甲酸酯预聚物3:将聚氧丙烯乙二醇(polyoxypropylene ethylene diol,agc社制
エクセノール
510,重均分子量4,000)与聚合mdi(東
ソー
社制
ミリオネート
mr-200)与碳二亚胺改性mdi(東
ソー
社制
ミリオネート
mtl)以nco/oh(摩尔比)成为35.0的方式混合,使混合物在80℃的条件下反应5小时而制造的氨基甲酸酯预聚物
[0224]
·
异氰脲酸酯体1:1,5-戊二异氰酸酯的异氰脲酸酯体
[0225]
·
异氰脲酸酯体2:1,6-己二异氰酸酯的异氰脲酸酯体
[0226]
·
脲基甲酸酯体1:1,5-戊二异氰酸酯的脲基甲酸酯体
[0227]
·
脲基甲酸酯体2:1,6-己二异氰酸酯的脲基甲酸酯体
[0228]
·
缩二脲体:1,6-己二异氰酸酯的缩二脲体
[0229]
·
硅烷偶联剂1:n-苯基-γ-氨基丙基三甲氧基硅烷
[0230]
·
硅烷偶联剂2:三乙氧基甲硅烷基硫丙基三甲氧基硅烷
[0231]
·
一元醇:正辛醇
[0232]
·
炭黑:親日化
カーボン
社制#200mp
[0233]
·
碳酸钙1:丸尾
カルシウム
社制
スーパー
s(重质碳酸钙)
[0234]
·
二氧化硅:日本
アエロジル
社制aerosil r972(表面处理热解法二氧化硅)
[0235]
·
脱水剂:
ソルベイジャパン
社制
ゼオシール
a-4(沸石(
ゼオラオイト
))
[0236]
·
增塑剂:
ジェイプラス
社制dinp(邻苯二甲酸二异壬酯)
[0237]
·
催化剂1:
サンアプロ
社制ucat-660m(dmdee(二吗啉代二乙基醚))
[0238]
·
特定多元醇1:旭硝子社制excenol450ed(聚氧丙烯四醇(eo末端),重均分子量500)
[0239]
·
特定多元醇2:日油社制
ユニオール
tg-330(聚氧丙烯-甘油基醚(三醇),重均分子量330)
[0240]
·
2官能多元醇1:三菱
ケミカル
社制ptmg-2000(聚丁二醇(二醇),重均分子量2,000)
[0241]
·
2官能多元醇2:三菱
ケミカル
社制14bg(1,4-丁二醇,分子量90)
[0242]
·
2官能多元醇3:日油社制
ユニオール
d-400(聚氧丙烯二醇(二醇),重均分子量400)
[0243]
·
2官能多元醇4:agc社制
エクセノール
3020(聚丙二醇(二醇),重均分子量3,000)
[0244]
·
2官能多元醇5:出光兴产社制poly bd r-15ht(液态聚丁二烯二醇,重均分子量1,200)
[0245]
·
2官能多元醇6:荒川化学工业社制d-6011(松香二醇,重均分子量1,000)
[0246]
·
3官能多元醇1:旭硝子社制preminol7001k(聚氧丙烯三醇(eo末端),重均分子量6,500)
[0247]
·
3官能多元醇2:agc社制
エクセノール
823(聚丙烯乙二醇(三醇),重均分子量5,000)
[0248]
·
碳酸钙2:丸尾
カルシウム
社制
カルファイン
200(用脂肪酸进行了表面处理的碳酸钙)
[0249]
·
滑石:日本
ミストロン
社制
ソープストン
a(平均粒径3.5~4.0μm,长宽比9.5)
[0250]
·
催化剂2:日东化成社制u-810(二辛基二月桂酸锡)
[0251]
·
催化剂3:sigma-aldrich社制dabco 33-lv(dabco 33%丙二醇溶液)
[0252]
〔叠层体的制造〕
[0253]
如下所述,制造出叠层体。
[0254]
<表面处理工序>
[0255]
通过对下述树脂基材1~3的表面实施下述表2~4所示的表面处理,从而分别获得了在各实施例和比较例中使用的表面处理后树脂基材各3种。
[0256]
例如,如果为实施例a1,则获得了实施了火焰处理3的树脂基材1、实施了火焰处理3的树脂基材2、和实施了火焰处理3的树脂基材3。
[0257]
(树脂基材)
[0258]
表面处理前的树脂基材1~3的详细内容如下所述。另外,树脂基材1~3都相当于结晶性热塑性树脂基材。
[0259]
·
树脂基材1:加有滑石的pp(加有滑石的聚丙烯)树脂基材(通过将
プライムポリマー
社制tsop6成型而获得的树脂基材)(表面自由能的极性项γ
p0
:0.2mjm-2
,表面自由能的分散项γ
d0
:30.2mjm-2
)(通过dsc测定而获得的吸热峰的起点温度:120℃)
[0260]
·
树脂基材2:pp-gf(加有玻璃纤维的聚丙烯)树脂基材(通过将日本
ポリプロ
社制
ファンクスター
成型而获得的树脂基材)(表面自由能的极性项γ
p0
:0.2mjm-2
,表面自由能的分散项γ
d0
:30.3mjm-2
)(通过dsc测定而获得的吸热峰的起点温度:123℃)
[0261]
·
树脂基材3:cfrtp(碳纤维增强热塑性树脂)树脂基材(表面自由能的极性项γ
p0
:0.6mjm-2
,表面自由能的分散项γ
d0
:30.7mjm-2
)(通过dsc测定而获得的吸热峰的起点温度:128℃)
[0262]
(表面处理)
[0263]
表面处理的详细内容如下所述。
[0264]
(1)火焰处理(火焰处理1~6)
[0265]
火焰处理使用arcogas社制的火焰处理装置(气体流量:3.7l/min(分钟),空气流量100l/min(分钟)),在表2~3所记载的条件(到基材的距离、处理速度、通过次数)下进行。
[0266]
这里,所谓处理速度,是火焰处理的速度,具体而言,是相对于基材移动火焰处理装置的速度[mm/sec(秒)]。此外,所谓到基材的距离,是火焰处理装置与基材的距离[mm]。此外,所谓通过次数,是扫过火焰的次数。例如,通过次数记载为“1”的是将火焰从基材的一端到另一端扫过1次的情况,通过次数记载为“3”的是将火焰从基材的一端到另一端扫过1次后再1次从另一端到一端扫过火焰,然后,再次,将火焰从基材的一端到另一端扫过的情况。
[0267]
(2)等离子体处理(等离子体处理1)
[0268]
等离子体处理使用等离子体处理装置,在表4所记载的条件(到基材的距离、处理速度、通过次数)下进行。
[0269]
这里,所谓处理速度,是等离子体处理的速度,具体而言,是相对于基材移动等离子体放电喷嘴的速度[mm/秒]。此外,所谓到基材的距离,是等离子体放电喷嘴与基材的距离[mm]。此外,所谓通过次数,是扫过等离子体放电喷嘴的次数。等离子体处理1由于通过次数为“1”,因此是将等离子体放电喷嘴(等离子体放电)从基材的一端到另一端扫过1次的情况。
[0270]
(3)uv处理(uv处理1~4)
[0271]
uv处理使用uv照射装置,在表4所记载的条件(到基材的距离、照射时间)下进行。
[0272]
这里,所谓到基材的距离,是uv照射装置与基材的距离[mm]。
[0273]
<固化前粘接剂层形成工序>
[0274]
在所得的各表面处理后树脂基材(宽度:25mm,长度:120mm,厚度:3mm)的一侧的表面整体,涂布如上所述获得的异氰酸酯系粘接剂,形成了固化前粘接剂层(厚度:3mm)。另外,在粘接剂为双组分型的情况下,以表1或表2~4的“主剂/固化剂(质量比)”栏所记载的比例混合。
[0275]
<贴合工序>
[0276]
进一步,在所形成的固化前粘接剂层上贴合相同种类的表面处理后树脂基材(宽度:25mm,长度:120mm,厚度:3mm)。
[0277]
例如,在实施例a1中,在实施了火焰处理3的树脂基材1上形成固化前粘接剂层的情况下,进一步,在所形成的固化前粘接剂层上贴合实施了火焰处理3的其它树脂基材1。
[0278]
此外,在将上述2块表面处理后树脂基材贴合时,在上述2块表面处理后树脂基材中以彼此对置的面整体大致重合的方式配置上述2块表面处理后树脂基材。
[0279]
这样操作而获得了依次具有树脂基材、固化前粘接剂层和树脂基材的叠层体前体。
[0280]
<固化工序>
[0281]
接着,通过将叠层体前体在23℃、50%rh的条件下静置3天,从而使固化前粘接剂层固化,获得了依次具有树脂基材、粘接剂层和树脂基材的叠层体(初始叠层体)。
[0282]
关于各实施例和比较例,分别获得了3种叠层体(初始叠层体)。例如,如果为实施例a1,则获得使用了树脂基材1作为表面处理前的树脂基材的叠层体、使用了树脂基材2作为表面处理前的树脂基材的叠层体、和使用了树脂基材3作为树脂基材的叠层体。
[0283]
<各种试验>
[0284]
使用如上所述获得的各初始叠层体进行以下各种试验。
[0285]
(耐水试验)
[0286]
进行了使如上所述获得的各初始叠层体在50℃的水中浸渍2周的耐水试验。在上述耐水试验后,将叠层体从水中拉起来,使其为耐水粘接性评价用叠层体。
[0287]
(耐热试验)
[0288]
进行了将如上所述获得的各初始叠层体在大气中、在90℃的条件下放置2周的耐热试验。使上述耐热试验的叠层体为耐热粘接性评价用叠层体。
[0289]
〔评价〕
[0290]
关于所得的各叠层体,如下所述进行了评价。
[0291]
(剥离试验)
[0292]
进行对如上所述获得的各初始叠层体(各样品数:10个)的粘接剂层用刀引入切口、在上述初始叠层体中将2块树脂基材彼此用手剥离的剥离试验,通过目视观察了上述剥离试验后的剥离面。
[0293]
关于如上所述获得的各耐水粘接性评价用叠层体、和各耐热粘接性评价用叠层体,也与上述同样地操作而进行了剥离试验。
[0294]
(剥离试验的评价基准)
[0295]
调查了对上述初始叠层体进行了的剥离试验后的剥离面中的破坏状态(具体而言,凝集破坏(cf)、树脂基材的材料破坏(mf)、界面剥离(af))、和上述各种破坏状态在上述
剥离面中所占的面积的比例(%)。
[0296]
将上述cf等破坏状态和该破坏状态在剥离面中所占的面积的比例的平均值(%)的结果示于表2~4的“破坏状态(初始粘接性)”栏。这里,作为上述结果的表示的一例的“cf100”表示破坏状态为凝集破坏,且上述凝集破坏的面积相对于剥离面的面积为100%。此外,“cf90af10”表示破坏状态以凝集破坏和界面剥离混合存在,且相对于剥离面的面积,凝集破坏的面积的比例为90%,界面剥离的面积的比例为10%。
[0297]
关于对上述耐水粘接性评价用叠层体进行了的剥离试验后的剥离面,进行了与上述同样的评价。将其结果示于表2~4的“破坏状态(耐水粘接性)”栏。
[0298]
关于对上述耐热粘接性评价用叠层体进行了的剥离试验后的剥离面,进行了与上述同样的评价。将其结果示于表2~4的“破坏状态(耐热粘接性)”栏。
[0299]
从粘接性的观点考虑,上述“初始粘接性”、“耐水粘接性”和“耐热粘接性”中都优选cf(包含mf)的比例高。此外,在实用上,在上述“初始粘接性”、“耐水粘接性”和“耐热粘接性”中都优选cf的比例超过0%。
[0300]
<70℃加热10分钟后的强度>
[0301]
所得的叠层体之中,关于使用了树脂基材1的初始叠层体(各样品数:10个),进行了在大气中、在70℃的条件下加热10分钟的加热试验。
[0302]
使用上述加热试验后的叠层体,按照jis k6850:1999进行拉伸试验(拉伸速度50mm/分钟,20℃的环境下),测定了剪切强度。将其结果示于表2~4的“70℃加热10分钟后的强度”栏。粘接耐久性优选值大的一方。
[0303]
[0304]
[0305][0306]
在表2~4中,处理时基材表面温度为表面处理时的树脂基材的表面温度。此外,树脂基材栏的“δ
p”和“δ
d”表示上述δ
p
和δd。测定方法如上所述。此外,树脂基材栏的“式(b)”表示上述式(b)所示的值。
[0307]
由表2~4可知,将式(a)为特定范围的异氰酸酯系粘接剂用于式(b)为特定范围的树脂基材的实施例都显示优异的粘接性(初始粘接性)、耐水粘接性和耐热粘接性。其中,式(a)为20~35,并且,式(b)为15以下的实施例a12~13、a16~17、a22、b3~4、c3~4和c9显示更优异的耐热粘接性。
[0308]
另一方面,式(a)和式(b)中的至少一者超出特定范围的比较例都是粘接性(初始粘接性)、耐水粘接性和耐热粘接性中的至少1者不充分(af100)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1