一种离子交换制备碲镉汞量子点的方法
1.本发明涉及一种离子交换制备碲镉汞(hg
x
cd
1-x
te)量子点的方法,属于量子点制备技术领域。
背景技术:
2.量子点作为零维半导体材料,具有比表面积大、表面活性高、可溶液加工、带隙可调、荧光量子效率高及多激子效应等特性,在光伏器件、医学成像、光通信、显示等领域具有良好的发展前景和应用价值。高质量量子点的合成是制备光电器件的前提,其中红外量子点包括ⅳ族(si,ge,gesn),
ⅴ
族(inas,insb),
ⅳ‑ⅵ
族(pbs,pbse,pbte),ⅵ族(hgcdte,hgse,hgte),
ⅰ‑ⅵ
族(ag2s, ag2se),
ⅰ‑ⅲ‑ⅵ
(agbis2,aginse2,cuins2,cuinse2)及钙钛矿型的cssni3, cssnpb
1-x
,fapbi3。这些红外量子点从短波红外(1.4-3μm)、中波红外(3-8μm) 覆盖至长波红外(8-15μm)的宽调谐性能,与可见光波段量子点相比,同样具备窄的荧光发射峰,较高的荧光量子效率,吸光能力强等优势,在光伏器件、医学成像、红外探测、场效应晶体管及红外发光二极管等多方面的应用潜力巨大。
3.赝二元化合物碲镉汞(hg
1-x
cd
x
te)半导体材料通过改变镉和汞的比例实现 1-3μm、3-5μm和8-14μm三个重要大气窗口的宽光谱红外光响应,响应速度快、探测率高和噪声低等优势,在红外探测器件上的应用具有十分重要的意义。制备碲镉汞半导体合金量子点,可以保持块体材料的优异的物理特性,同时通过精准调控汞和镉的比例实现cdte量子点荧光光谱从可见光到近红外光谱波段的调控,也可以进一步拓宽碲镉汞材料在柔性器件上的应用。
4.1993年bawendi等人提出利用快速注入有机金属前驱体使之达到过饱和快速完成成核及生长的可控制备量子点的方法(j.am.chem.soc,1993, 115(19):8706-8715.),其中镉基量子点的相关研究较为广泛,自上世纪九十年代起对于如何提升镉基量子点的荧光量子产率及如何修饰量子点表面缺陷等进行了深入研究,但对于碲镉汞量子点的可控制备报道相对较少。andrew m.smith 等人首先利用高温热注入法得到cdte量子点,随后利用阳离子交换法将部分镉阳离子替换为汞阳离子从而得到碲镉汞量子点。该方法通过在300℃高温下得到cdte量子点,此方法虽然能得到单分散性的碲镉汞量子点,但并不适用于工业化生产需求(j.am.chem.soc.2011,133(1):24-26.)。wang hongzhe等人通过替换镉源的包覆剂及汞源,同样是采用先高温热注入得到cdte量子点,随后通过阳离子替换得到碲镉汞量子点(dalton trans.2012,41(41):12726-12732.)。制备方法复杂且采用高温热注入方法,在一定程度上限制了量子点在相应领域的实际应用。因此,如何设计和制备胶体cdte量子点,降低热注入成核温度并维持碲镉汞量子点良好的单分散性十分必要。
技术实现要素:
5.本发明解决的技术问题是:现有制备hg
x
cd
1-x
te量子点的方法存在热注入温度高、制备得到的量子点分散性差等问题。
6.为了解决上述技术问题,本发明提供了一种离子交换制备hg
x
cd
1-x
te量子点的方法,包括以下步骤:
7.步骤1:以氧化镉或硬脂酸镉作为镉源,在氮气氛围保护下利用油酸溶解并分散在1-十八烯中,在100-150℃下搅拌溶解镉源得到澄清溶液,然后加热至 300℃维持30min,随后降温至250℃,得到镉前驱体溶液;以碲粉作为碲源,利用三辛基膦溶解并分散在1-十八烯中,在80-160℃下溶解碲源得到澄清溶液随后冷却至室温,得到碲前驱体溶液;将所述的碲前驱体溶液注入到镉前驱体溶液中并维持生长时间为0.5-10min,得到不同发射光峰位的cdte量子点;将所制备的cdte量子点依次分散在甲醇、乙醇中进行离心后,分散于氯仿中,得到cdte量子点溶液;
8.步骤2:以碘化汞或氯化汞作为汞源,将其分散在含有氢氧化钾及烷基硫醇的甲醇溶液中,利用烷基硫醇溶解汞源得到烷基硫醇汞白色沉淀,用甲醇及乙醚洗涤后离心,真空干燥后用氯仿溶解,得到汞前驱体溶液;
9.步骤3:在氮气保护作用下,在步骤1所得的cdte量子点溶液中加入辛胺于30-60℃加热,得到cdte前驱体溶液,将步骤2所得的汞前驱体同样在 30-60℃加热,随后将汞前驱体溶液注入到cdte前驱体溶液中,进行离子交换反应1-10min,随后分散在甲醇中离心得到hg
x
cd
1-x
te量子点。
10.优选地,所述步骤1中的镉源和碲源的摩尔比为1~5:1,所述镉源和油酸的摩尔比为1:2~4,所述镉前驱体溶液的浓度为0.01-0.1m。
11.优选地,所述步骤1中的碲源和三辛基膦的摩尔比为1:2~4,所述碲前驱体溶液的浓度为0.01-0.1m。
12.优选地,所述步骤1中的cdte量子点溶液的浓度为0.005~0.05m。
13.优选地,所述步骤2中的汞源、氢氧化钾及烷基硫醇摩尔比为1:2~5:2~5,所述烷基硫醇为辛硫醇、十二烷基硫醇、十六烷基硫醇及十八烷基硫醇中的至少一种。
14.优选地,所述步骤2中的汞前驱体溶液的浓度为0.01~0.05m。
15.优选地,所述步骤3中的cdte量子点溶液与辛胺的体积比为10:0.5~2。
16.优选地,所述步骤3中的cdte前驱体溶液与汞前驱体溶液的配比按照cd: hg的摩尔比为1:0.1~0.3的比例进行计算。
17.本发明与现有技术相比,具有如下有益效果:
18.1.本发明利用通过降低成核温度的热注入方法先制备得到cdte量子点,并将其分散在含有烷基硫醇作为配体的汞前驱体的氯仿溶液中,随后在温和条件下通过离子交换制备得到hg
x
cd
1-x
te量子点;相比于高温热注入法,本发明提供的反应实现条件较温和,反应温度较低,操作简单,可实现规模化生产;
19.2.本发明的方法可获得高质量的hg
x
cd
1-x
te量子点,其具有较均匀的粒径分布和高的单分散性,在近红外光电探测应用领域具有重要意义。
附图说明
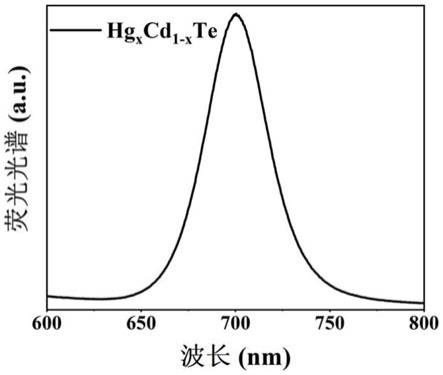
20.图1为本发明中实施例1中所制备的hg
x
cd
1-x
te量子点的荧光发射图;
21.图2为本发明中实施例3中所制备的hg
x
cd
1-x
te量子点的tem图;
22.图3为本发明中实施例4中所制备的hg
x
cd
1-x
te量子点的hrtem图。
具体实施方式
23.为使本发明更明显易懂,兹以优选实施例,并配合附图作详细说明如下。
24.实施例1
25.一种离子交换制备hg
x
cd
1-x
te(x=0~1)量子点的方法,包括以下步骤:
26.在氮气保护作用下,称取2mmol(0.256g)的氧化镉,5ml油酸,45ml 1
‑ꢀ
十八烯在120℃下搅拌溶解得到澄清浅黄色溶液并维持1h,随后将其加热至300℃并维持30min,随后降温至250℃。称取1mmol(0.127g)碲粉,4ml三辛基膦,11ml的1-十八烯在140℃下溶解得到澄清浅绿色溶液,随后冷却至室温。将碲前驱体溶液快速注入到镉前驱体溶液中控制生长时间为1.5min,将其用甲醇、乙醇离心,随后将其分散入100ml氯仿溶液中。将0.4mmol (0.0404g)十二烷基硫醇,0.4mmol(0.0111g)氢氧化钾分散入10ml甲醇中,随后加入0.2mmol(0.0908g)碘化汞搅拌12h,将其抽滤,用甲醇和乙醚洗涤后在8000rpm下离心5min,将所得白色沉淀在真空干燥箱内干燥12h,随后将其分散入10ml氯仿溶液中。在氮气保护下,分散入氯仿溶液中的cdte量子点取10ml,随后加入0.5ml辛胺并在50℃下加热。分散在氯仿中的汞前驱体溶液在50℃下加热,随后将10ml汞前驱体溶液注入到cdte量子点溶液中,控制汞阳离子交换时间为4min,随后利用甲醇将其离心分散入正己烷内,得到 hg
x
cd
1-x
te量子点。
27.对实施例1制备得到的hg
x
cd
1-x
te量子点进行荧光测试,如图1所示, hg
x
cd
1-x
te量子点的荧光发射峰峰位在700.2nm。
28.实施例2
29.一种离子交换制备hg
x
cd
1-x
te(x=0~1)量子点的方法,包括以下步骤:
30.在氮气保护作用下,称取2mmol(1.3587g)的硬脂酸镉,4ml油酸,45ml 1-十八烯在150℃下搅拌溶解得到澄清浅黄色溶液并维持1h,随后将其加热至 300℃并维持30min,随后降温至250℃。称取2mmol(0.254g)碲粉,4ml三辛基膦,6ml 1-十八烯在120℃下溶解得到澄清浅绿色溶液,随后冷却至室温。将碲前驱体溶液快速注入到镉前驱体溶液中控制生长时间为1min,将其用甲醇、乙醇离心,随后将其分散入100ml氯仿溶液中。将0.4mmol(0.0584g) 辛硫醇,0.4mmol(0.0111g)氢氧化钾分散入10ml甲醇中,随后加入0.3mmol (0.1362g)碘化汞搅拌24h,将其抽滤,用甲醇和乙醚洗涤后在8000rpm下离心5min,将所得白色沉淀在真空干燥箱内干燥12h,随后将其分散在10ml 氯仿溶液中。在氮气保护下,将分散入氯仿溶液中的cdte量子点取10ml,随后加入0.5ml辛胺并于45℃下加热,分散在氯仿中的汞前驱体溶液于45℃下加热,随后将10ml汞前驱体溶液注入到cdte量子点溶液中,控制汞阳离子交换时间为10min,随后利用甲醇将其离心分散入正己烷内,得到hg
x
cd
1-x
te量子点。
31.实施例3
32.一种离子交换制备hg
x
cd
1-x
te(x=0~1)量子点的方法,包括以下步骤:
33.在氮气保护作用下,称取2mmol(0.256g)的氧化镉,5ml油酸,45ml 1
‑ꢀ
十八烯在100℃下搅拌溶解得到澄清浅黄色溶液并维持1h,随后将其加热至 300℃并维持30min,随后降温至250℃。称取1mmol(0.127g)碲粉,4ml三辛基膦,11ml 1-十八烯在160℃下溶解得到澄清浅绿色溶液,随后冷却至室温。将碲前驱体溶液快速注入到镉前驱体溶液中控制生长时间为1.5min,将其用甲醇、乙醇离心,随后将其分散入100ml氯仿溶液中。将0.4mmol
(0.1034g) 十六烷基硫醇,0.4mmol(0.0111g)氢氧化钾分散入10ml甲醇中,随后加入 0.2mmol(0.0542g)氯化汞搅拌6h,将其抽滤,用甲醇和乙醚洗涤后在8000 rpm下离心5min,将所得白色沉淀在真空干燥箱内干燥24h,随后将其分散在10ml氯仿溶液中。在氮气保护下,将分散入氯仿溶液中的cdte量子点取 10ml,随后加入0.5ml辛胺并于60℃下加热,分散在氯仿中的汞前驱体溶液于60℃下加热,随后将10ml汞前驱体溶液注入到cdte量子点溶液中,控制汞阳离子交换时间为10min,随后利用甲醇将其离心分散入正辛烷内,得到 hg
x
cd
1-x
te量子点。
34.对实施例3制备得到的hg
x
cd
1-x
te量子点进行结构表征,如图2所得的透射电镜图可以看出,制备出的hg
x
cd
1-x
te量子点粒径均匀,且单分散性好, hg
x
cd
1-x
te量子点的粒径大小约4.2nm。
35.实施例4
36.一种离子交换制备hg
x
cd
1-x
te(x=0~1)量子点的方法,包括以下步骤:
37.在氮气保护作用下,称取2mmol(0.256g)的氧化镉,5ml油酸,45ml 1
‑ꢀ
十八烯在120℃下搅拌溶解得到澄清浅黄色溶液并维持1h,随后将其加热至 300℃并维持30min,随后降温至250℃。称取1.5mmol(0.1905g)碲粉,6ml 三辛基膦,9ml 1-十八烯在120℃下溶解得到澄清浅绿色溶液,随后冷却至室温。将碲前驱体溶液快速注入到镉前驱体溶液中控制生长时间为2min,将其用甲醇、乙醇离心,随后将其分散入100ml氯仿溶液中。将0.6mmol(0.1719g) 十八烷基硫醇,0.6mmol(0.0168g)氢氧化钾分散入10ml甲醇中,随后加入 0.2mmol(0.0908g)碘化汞搅拌12h,将其抽滤,用甲醇和乙醚洗涤后在5000 rpm下离心10min,将所得白色沉淀在真空干燥箱内干燥12h,随后将其分散在10ml氯仿溶液中。在氮气保护下,将分散入氯仿溶液中的cdte量子点取 10ml,随后加入1ml辛胺并于50℃下加热,分散在氯仿中的汞前驱体溶液在 50℃下加热,随后将10ml汞前驱体溶液注入到cdte量子点溶液中,控制汞阳离子交换时间为6min,随后利用甲醇将其离心分散入甲苯内,得到hg
x
cd
1-x
te 量子点。
38.对实施例4制备得到的hg
x
cd
1-x
te量子点进行结构表征,如图3所示,制备得到的hg
x
cd
1-x
te量子点同样呈现单分散性,且粒子的晶格间距为
39.实施例5
40.一种离子交换制备hg
x
cd
1-x
te(x=0~1)量子点的方法,包括以下步骤:
41.在氮气保护作用下,称取2mmol(0.256g)的氧化镉,5ml油酸,45ml 1
‑ꢀ
十八烯在150℃下搅拌溶解得到澄清浅黄色溶液并维持1h,随后将其加热至 300℃并维持30min,随后降温至250℃。称取0.4mmol(0.0510g)碲粉,2ml 三辛基膦,13ml 1-十八烯在150℃下溶解得到澄清浅绿色溶液,随后冷却至室温。将碲前驱体溶液快速注入到镉前驱体溶液中控制生长时间为1.5min,将其用甲醇、乙醇离心,随后将其分散入100ml氯仿溶液中。将0.6mmol(0.0607g) 十二烷基硫醇,0.6mmol(0.0168g)氢氧化钾分散入10ml甲醇中,随后加入0.3mmol(0.1362g)碘化汞搅拌12h,将其抽滤,用甲醇和乙醚洗涤后在 8000rpm下离心5min,将所得白色沉淀在真空干燥箱内干燥8h,随后将其分散在10ml氯仿溶液中。在氮气保护下,将分散入氯仿溶液中的cdte量子点取10ml,随后加入0.5ml辛胺并于50℃下加热,分散在氯仿中的汞前驱体溶液在50℃下加热,随后将10ml汞前驱体溶液注入到cdte量子点溶液中,控制汞阳离子交换时间为10min,随后利用甲醇将其离心分散入正己烷内,得到
hg
x
cd
1-x
te量子点。
42.以上所述,仅为本发明的较佳实施例,并非对本发明任何形式上和实质上的限制,应当指出,对于本技术领域的普通技术人员,在不脱离本发明的前提下,还将可以做出若干改进和补充,这些改进和补充也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1