一种碳点以及碳点基复合材料的制备方法
1.本发明属于碳点制备技术领域,具体涉及一种碳点以及碳点基复合材料的制备方法。
背景技术:
2.刺激响应型光学材料因其在光电设备、生物成像、显示设备、传感器等领域中应用前景广阔而受到越来越多的关注,特别是在信息加密领域,开发新颖的刺激响应型智能材料对于避免模仿的防伪技术极为重要。室温磷光(rtp)材料因其长寿命磷光、大的斯托克斯位移和小背景干扰而受到广泛关注,有望成为该领域有前景的候选材料。为了实现有效的rtp,通常需要有效的自旋轨道耦合来实现单重态到三重态的系间跨越(isc)和稳定的三重激发态。但是由于三重激发态的自旋禁止跃迁特征和非辐射跃迁过程导致的三重态激子失活,很多rtp材料都面临磷光寿命短、量子产率低、需要严格脱氧的情况以及与可印刷油墨不相容的问题。报道最多的rtp材料,例如有机金属和纯有机化合物,也存在成本高、稳定性差、制备复杂以及毒性高等缺点。
3.碳点(cds),一种尺寸在10nm以内的零维碳材料,具有低毒性、良好的生物相容性和优异的光学性能,并且易于制备可用于大规模生产,因此在生物传感器、太阳能电池、发光二极管和催化领域得到了广泛的应用。近年来,许多学者已经广泛探索了碳点的荧光性质,但是关于其rtp特性的报道却非常有限。同样是由于不稳定的三重激发态物质、氧诱导的磷光猝灭和无效的系间跨越(isc)等问题,开发室温磷光cds仍面临巨大挑战。
4.关于cds余辉的第一次报道可以追溯到2012年,当时lin等人使用pb
2+
使荧光cds发出强而稳定的室温磷光,但是这种重原子辅助方法是有毒的,因此很少受到关注。2013年,deng等人报道了聚乙烯醇(pva)基质中碳点的磷光现象,磷光归因于碳点表面的c = o键,并且具有很长的寿命(380 ms)。这是cds第一次无需重原子掺杂即可获得的磷光现象,从那时起,cds的余辉正式进入研究人员的视野。
5.cds要实现余辉排放,有两个关键的先决条件:(1)cds应当具有合适的能级结构以产生三重态激子进行电子跃迁,并且在s0和t1之间具有较小的δest以实现有效的isc;(2)必须有可以保护cd产生的三重态激子免于非辐射失活的特定结构。cds中余辉发射的起源主要归因于c = o / c = n相关的官能团,这些官能团表现出很强的自旋轨道耦合能力,可以有效地产生三重态激子。因此,通过将n、p等杂原子掺杂到cds中,可以很好地满足第一个条件。杂原子掺杂不仅可以极大地促进自旋-轨道耦合,而且可以在cds上产生丰富的表面状态,从而使它们产生有效的isc过程。为了满足保护三重态激子免于非辐射跃迁的第二个必要条件,可以设计和构建基质辅助系统和自保护结构来固定cds,从而产生高效的rtp发射。cds在聚合物和有机化合物等有机基质中的余辉主要归因于氢键的形成,它可以抑制分子内振动并稳定三重态,从而产生余辉发射。不同无机基质中cds的余辉机理完全不同,并且高度取决于cds与不同基质之间的多种相互作用(例如刚性结构共价键、结构限制、能量转化等)。自保护cds始终具有类似聚合物的结构,可以用作刚性基质来激活余辉发射。
6.近年来,已报道的用于构建基于cds的rtp材料的主要形式有两种:一种是基质辅助系统,该系统将cds固定在刚性基质中,并通过基质涂层形成淬灭剂阻挡层,从而产生高效的rtp发射;另一种是cds的自我保护性结构,即cds的表面带有许多聚合物链,或者cds具有类似聚合物的结构,可作为刚性基质来保护它们免受外部环境干扰,从而获得rtp发射。
7.现已报道的一些涉及刺激响应性磷光材料由于寿命短、需要严格的脱氧条件或与制造可印刷油墨不兼容,实际上不适合用于信息加密和防伪的常规应用。因此,我们期望开发出具有长寿命的室温磷光(rtp)材料,但由于三重激发态的自旋禁止跃迁特征,制造此类材料非常具有挑战性。
技术实现要素:
8.本发明的目的在于提供一种碳点以及碳点基复合材料的制备方法。
9.为实现上述目的,本发明采取的技术方案如下:一种碳点的制备方法,制备步骤如下:(a)、将乙二胺四乙酸四钠、naoh置于1#容器中,加水溶解,随后用微波炉加热1#容器至有黑色碳化块生成,取出1#容器放至室温;(b)、去除1#容器中的黑色碳化块、保留白色与褐色相间的固体,向1#容器中加入水,待固体全部溶解后离心,弃去沉淀,保留上清液,冻干,得到碳点粉末;其中,原料用量配比为乙二胺四乙酸四钠∶naoh=(0.5-1.5)g∶(0.3-1.0)g。
10.较好地,原料用量配比为乙二胺四乙酸四钠∶naoh∶步骤(a)中的水∶步骤(b)中的水=(0.5-1.5)g∶(0.3-1.0)g∶(10-50)ml∶(10-20)ml。
11.较好地,步骤(a)和步骤(b)中,在超声条件下溶解。
12.较好地,步骤(a)中,微波炉功率为560-700 w、微波炉加热100-150 s。
13.较好地,步骤(a)中,微波炉加热过程中每隔15-20 s取出1#容器摇匀溶液。
14.一种碳点基复合材料的制备方法,制备步骤如下:(a)、将乙二胺四乙酸四钠、naoh置于1#容器中,加水溶解,随后用微波炉加热1#容器至有黑色碳化块生成,加热完成后取出1#容器放至室温;(b)、去除1#容器中的黑色碳化块、保留白色与褐色相间的固体,向1#容器中加入水,待固体全部溶解后离心,弃去沉淀,保留上清液即碳点溶液;(c)、将聚合物置于2#容器中,按质量体积比聚合物∶水=(0.2-0.5)g∶(25-50)ml加水,分散,得分散液;所述聚合物为聚乙烯醇pva或聚丙烯酰胺pam;(d)、将步骤(b)所得碳点溶液和步骤(c)所得分散液按体积比(2-8)∶5置于3#容器中混合均匀;随后将混合液用微波炉加热3#容器至溶液蒸干,放至室温,加水(10-20ml)溶解,冻干,得到碳点基复合材料;其中,原料用量配比为乙二胺四乙酸四钠∶naoh=(0.5-1.5)g∶(0.3-1.0)g。
15.较好地,原料用量配比为乙二胺四乙酸四钠∶naoh∶步骤(a)中的水∶步骤(b)中的水=(0.5-1.5)g∶(0.3-1.0)g∶(10-50)ml∶(10-20)ml。
16.较好地,步骤(a)和步骤(b)中,在超声条件下溶解;步骤(c)中,在超声条件下分散;步骤(d)中,在超声条件下混合均匀。
17.较好地,步骤(a)和步骤(d)中,微波炉功率为560-700 w;步骤(a)中微波炉加热
100-150 s;步骤(d)中微波炉加热80-120 s。
18.较好地,步骤(a)和步骤(d)中,微波炉加热过程中每隔15-20 s取出1#容器或3#容器摇匀溶液。
19.有益效果:本发明采用绿色无污染的乙二胺四乙酸四钠为合成碳点的原料,通过加入适量的氢氧化钠,通过用微波炉加热一定时间就可以得到碳点,这个合成过程极其简单且易操作;再分别向得到的碳点溶液中加入聚乙烯醇或聚丙烯酰胺进行微波加热,得到碳点基复合材料(包覆的碳点),该碳点基复合材料具有rtp特性、长余辉发射特性,可在光电设备、生物成像、显示设备、传感器等领域中应用。
附图说明
20.图1为实施例1制备的cds的tem图(a)和hr-tem图(b)。
21.图2为实施例1制备的cds的afm图(a、b)。
22.图3为实施例2制备的cds@pva的xrd图。
23.图4为实施例1-3制备的cds(a)、cds@pva(b)、cds@pam(c)的磷光强度光谱。
24.图5 为采用不同的激发波长激发实施例1制备的cds的荧光光谱。
25.图6 为采用激发波长285和345 nm分别激发实施例1制备的cds的磷光强度光谱。
26.图7 为采用不同的激发波长激发实施例1制备的cds的磷光强度光谱。
27.图8 为采用激发波长345 nm激发实施例1-2制备的cds和5cds@5pva的磷光寿命图谱。
28.图9 为实施例1制备的cds的磷光衰减照片:a-激发波长254nm,b-激发波长365nm。
29.图10 为实施例1-3制备的cds、5cds@5pva、5cds@5pam 的ft-ir。
30.图11 为实施例1-3制备的cds、5cds@5pva、5cds@5pam的紫外可见漫反射光谱(uv vis drs)。
31.图12 为实施例1制备的cds的x射线光电子能谱分析(xps):a-总谱图,b-c1s的精细谱,c-n1s的精细谱,d-o1s的精细谱。
32.图13为345 nm激发下实施例2制备的5cds@5pva的变温磷光测试图谱。
33.图14为285nm激发下实施例2制备的5cds@5pva的蓝紫色磷光变温寿命图谱。
34.图15为330nm激发下实施例2制备的5cds@5pva的绿色磷光变温寿命图谱。
具体实施方式
35.为使本发明更加清楚、明确,以下对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
36.实施例1
‑‑
碳点(cds)的制备在分析天平上称取0.5 g乙二胺四乙酸四钠(edta
· 4na)和0.3 g naoh,转移至小烧杯中,加入10 ml去离子水,超声3 min至固体全部溶解;随后用微波炉(功率700 w)加热小烧杯 120 s至有黑色碳化块生成(在生成黑色碳化块之前烧杯已经没有水了;加热过程中每隔15 s取出烧杯摇匀溶液),加热完成后取出烧杯放至室温;用镊子去除黑色碳化块,烧杯中呈现白色固体与褐色固体相间的状态,加入10 ml水,超声3 min至固体全部溶解,转移至离心管,放入离心机离心15 min,弃去沉淀,保留上清液,即cds溶液,冻干得到
cds粉末。
37.实施例2
‑‑
碳点基复合材料cds@pva的制备称取0.2 g聚乙烯醇(pva)固体于小烧杯中,加入25 ml水溶解超声3 min得到pva分散液,待用;将实施例1中得到的cds溶液分为2 ml、5 ml和8 ml于三个烧杯中,每份加入5 ml pva分散液,超声3 min至混合均匀;随后将三份溶液分别用微波炉(功率700 w)加热100 s至溶液蒸干(加热过程中每隔15 s取出烧杯摇匀溶液),加热完成后取出烧杯放至室温,加入10 ml水溶解,冻干,得到碳点基复合材料cds@pva粉末,与2 ml、5 ml和8 ml cds溶液对应的碳点基复合材料cds@pva粉末分别标记为:2cds@5pva、5cds@5pva、8cds@5pva。
38.实施例3
‑‑
碳点基复合材料cds@pam的制备与实施例1的不同之处在于:将实施例2中的聚乙烯醇(pva)替换为聚丙烯酰胺pam,得到的产物为碳点基复合材料cds@pam粉末,与2 ml、5 ml和8 ml cds溶液对应的碳点基复合材料cds@pam粉末分别标记为:2cds@5pam、5cds@5pam、8cds@5pam。
39.实验分析测试:图1为实施例1制备的cds的tem图(a)和hr-tem图(b)。由图1可以看出:碳点存在,碳点的尺寸在1-10 nm,晶格条纹取向不一、显现交叉状。
40.图2为实施例1制备的cds的afm图(a、b)。由图2可以看出:碳点的厚度为0.1-6 nm。
41.图3为实施例2制备的cds@pva的xrd图。由图3可以观察到属于碳的特征峰。
42.图4为330 nm激发波长下实施例1-3分别制备的cds(a)、cds@pva(b)、cds@pam(c)的磷光强度光谱。由图4a可知:原料的微弱磷光得到了极大增强,cds粉末在330 nm激发波长下发出519 nm的绿色磷光;由图4b可知:不同含量cds溶液与pva的复合对比中,包覆cds含量适中的5cds@5pva表现出了更好的磷光特性;由图4c可知:不同含量cds溶液与pam复合对比中,5cds@5pam与8cds@5pam磷光强度较为相近。
43.图5为采用不同的激发波长激发实施例1制备的cds的荧光光谱。由图5可知:cds的发射波长没有发生明显移动。
44.图6为采用激发波长285和345 nm分别激发实施例1制备的cds的磷光强度光谱。由图6可知:激发波长285和345 nm分别对应有蓝紫色磷光发射(em1)以及绿色磷光发射(em2)。
45.图7为采用不同的激发波长激发实施例1制备的cds的磷光强度光谱。由图7可以观察到明显的磷光激发依赖性。
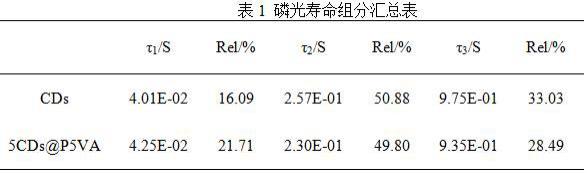
46.图8为采用激发波长345 nm激发实施例1-2制备的cds和5cds@5pva的磷光寿命图谱。由图8可知:cds和5cds@5pva的磷光寿命没有明显差别,采用三指数方程拟合得到二者的平均磷光寿命为0.75s和0.70 s,每部分的百分含量如表1所示。
47.图9为实施例1制备的cds的磷光衰减照片:a-激发波长254 nm,b-激发波长365nm。由图9a可知:254 nm激发下,蓝紫色余晖可达2 s;由图9b可知:365 nm激发下,绿色余晖可达7s。
48.图10为实施例1-3制备的cds、5cds@5pva、5cds@5pam的ft-ir。通过与原料进行对比,我们可以看出,这三种样品具有相类似的官能团振动,存在羟基、羧基、c-n等,但是在后续合成的cds 以及包覆不同聚合物的5cds@5pva、5cds@5pam 中羧基两个吸收峰也有所保留,由此可推及微波加热过程中没有发生大程度的脱羧反应,碳化程度较弱。而c-n的变化则是由于反应过程中大量的n会以气体的形式离去。
49.图11为实施例1-3制备的cds、5cds@5pva、5cds@5pam的紫外可见漫反射光谱(uv vis drs)。从cds紫外可见漫反射光曲线可知:微波加热过程中没有发生大程度的脱羧反应,碳化程度较弱,加热过程中部分n元素以nh3等可能的形式流失到空气中;从5cds@5pva、5cds@5pam紫外可见漫反射光曲线可知:pva或者pam包覆碳点的吸收峰相对于cds的吸收峰有明显的红移,认为是在包覆聚合物的过程中进行了二次微波加热,导致了碳点碳化程度的进一步增加。
50.图12为实施例1制备的cds的x射线光电子能谱分析(xps):a-总谱图,b-c1s的精细谱,c-n1s的精细谱,d-o1s的精细谱。由edta的分子式可知:相较于edta中n元素的百分含量,cds中的n元素含量减少,进一步推断微波加热过程中n元素以nh3等可能的形式流失到空气中。
51.为了排除长余辉来源于热延迟荧光(tadf)的可能,我们利用345 nm激发光源对5cds@5pva做了变温磷光测试,结果如图13所示,由于温度的升高使得三重态激子的振动转动等非辐射跃迁过程变得剧烈,导致三重态激子失活,磷光强度变弱。随后我们还分析了5cds@5pva温度相关的瞬态光致发光衰减过程,285nm激发下的蓝紫色磷光的余辉强度如图14所示,330nm激发下的黄绿色磷光的余辉强度如图15所示,在衰减过程中随着温度的升高而降低,这是室温磷光的特征,因为随着温度的升高非辐射失活的速率常数变大。与tadf的特性不同,三重态激子振动和转动过程增强使三重态激子通过逆系间跨越过程回到单重态能级的可能性随温度升高而增加,热活化能活化余晖,因此延迟成分的比例增加。
52.有了以上数据作为支撑,我们可以进一步确定磷光的来源,由tem下的形貌可以看出碳点呈现聚集交联状态,edta 4na原料在强碱性条件下加速碳化,形成了一定程度的石墨化共轭结构,呈紧密的碳核态,共轭骨架周围的烷基链、羧酸根以氢键、静电力等相互作用进一步交联,少量的cds聚集交联成簇,在复合上pva/pam后,碳簇以氢键、静电力等相互作用分散、嵌入、交联在pva/pam的网状结构中。从ft-ir、uv vis drs以及xps对碳点结构的分析中可以看出c=o/c-o/c-n的n
→
π*跃迁是磷光的主要来源,碳点的交联聚集以及pva/pam的网状结构为其提供了一个刚性的环境,限制了非辐射跃迁过程,保护了三重态激子,并且对于嵌入pva/pam的碳簇还起到了良好的氧阻隔作用,避免了氧猝灭过程。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1