硼硫掺杂碳量子点及其制备方法和应用
1.本发明涉及荧光材料技术领域,特别涉及硼硫掺杂碳量子点及其制备方法和应用。
背景技术:
2.比率荧光传感器是荧光传感器的一种,大多数比率荧光传感器是由双发射峰组成,依据两个发射峰荧光强度的升降变化可实现对目标分析物的定性和定量检测。与单峰发射的荧光传感器相比,比率型荧光传感器因其自我校正功能因而具有更好的检测准确性。比率型荧光传感器的出现解决了只有单个发射波长的荧光传感器易受到激发光源波动、检测环境以及探针局部浓度变化问题。至今为止,比率型荧光传感器的构建依赖于将两种不同的发光纳米粒子通过化学/物理的方法进行结合,但这种方法存在两种纳米粒子分布不均,纳米粒子尺寸小,难以包埋等等缺点。
3.碳量子点,又称为碳点(carbon dots,cds)是一种尺寸在1~10nm之间的新型荧光纳米材料,自2004年被发现以来一直受到广泛的关注。与其他传统的量子点相比,碳点有以下优点:合成方法简单、合成原料来源广、环保、可持续的优点;由于其尺寸小且表面具有丰富的含氧官能团如氨基,羧基及羰基等,这也使其具有较好的分散性,有利于在水溶液中的检测;其毒性低,生物相容性好,可作用于生物体内检测;还具有较好的光漂白性,在连续数小时的激光照射下,仍能够保持良好的荧光稳定性,因而碳量子点常被应用于荧光传感器的构建以及应用。
4.恩诺沙星(enr)属于第三代喹诺酮类抗生素,其常被用作动物用药以治疗动物呼吸道及胃肠道感染;enr还具有广谱的抑菌效果,对革兰氏阳性菌、阴性菌以及霉形体具有明显的抑菌作用。目前,enr的常用检测方法有高效液相色谱法、质谱法、电泳法以及电分析化学法。但这些方法仍存在一定限制,高效液相色谱法与质谱检测法虽然具备灵敏、检测结果准确的优点但其所需仪器昂贵、检测速度较慢;电泳法及电化学法易受到其他物质干扰,因此开发一种简便、快速、灵敏的enr检测方法具有重要意义。
技术实现要素:
5.本发明旨在至少解决现有技术中存在的技术问题之一。为此,本发明的第一方面是提供一种硼硫掺杂碳量子点的制备方法,该方法制的硼硫掺杂碳量子点能够作为比率荧光传感器,对抗生素恩诺沙星进行特异性检测。
6.本发明的第二方面是提供上述制备方法得到的硼硫掺杂碳量子点。
7.另外,本发明还提供所述硼硫掺杂碳量子点的应用。
8.具体地,本发明采取如下的技术方案:
9.本发明的第一方面是提供一种硼硫掺杂碳量子点的制备方法,包括如下步骤:使三氨基苯硼酸或其衍生物和硫脲的酸性混合溶液进行水热反应,得到硼硫掺杂碳量子点;所述酸性混合溶液的ph为0.35~6;所述水热反应的温度为150~200℃,所述水热反应的时
间为3~10h。
10.本发明通过采用三氨基苯硼酸或其衍生物作为原料,可同时作为碳源和硼源,且三氨基苯硼酸具有苯环,有利于碳量子点共轭结构的产生,与硫脲在酸性条件下进行反应后可制得特殊的硼硫掺杂碳量子点,该硼硫掺杂碳量子点具有优异的荧光性能,具有特殊的发射波长,能够对作为比率荧光传感器对抗生素恩诺沙星进行特异性检测。
11.在本发明的一些实例中,所述酸性混合溶液的ph为0.35~5.5,优选0.5~5,更优选0.35~2,再优选例如0.35、0.5、1、1.5、2、2.5、3、3.5、4、4.5、5、5.5等。酸性条件对原料的溶解以及硼硫掺杂碳量子点的性能具有重要影响,在非酸性条件下得到的碳量子点的荧光发射波长会发生改变,从而影响其特异选择性。
12.在本发明的一些实例中,通过将三氨基苯硼酸或其衍生物和硫脲溶于酸中得到所述酸性混合溶液,所述酸包括硝酸、盐酸中的至少一种。
13.在本发明的一些实例中,所述酸的浓度为0.05~0.4m,优选0.1~0.4m,例如0.05m,0.1m,0.2m,0.3m,0.4m等。
14.在本发明的一些实例中,所述水热反应的温度为160~190℃,优选175~185℃,例如150℃、155℃、160℃、165℃、170℃、175℃、180℃、185℃、190℃、195℃、200℃等。
15.在本发明的一些实例中,所述水热反应的时间为3~8h,优选3~6h,更优选3~5h,例如3h、4h、5h、6h、7h、8h、9h、10h等。
16.在本发明的一些实例中,所述三氨基苯硼酸衍生物包括三氨基苯硼酸的水合物、盐中的至少一种。
17.在本发明的一些实例中,所述三氨基苯硼酸或其衍生物中的硼元素与硫脲中的硫元素的摩尔比为1:0.8~1.5,优选1:1~1.2,例如1:0.8,1:0.9,1:1,1:1.1,1:1.2,1:1.3,1:1.4,1:1.5等。
18.在本发明的一些实例中,所述三氨基苯硼酸衍生物为三氨基苯硼酸一水合物,所述三氨基苯硼酸一水合物与硫脲的质量比为1~3:1,优选1.5~2:1,更优选1.6~1.9:1,例如1:1,1.2:1,1.3:1,1.4:1,1.5:1,1.6:1,1.7:1,1.8:1,1.9:1,2:1,2.5:1,3:1等。
19.在本发明的一些实例中,所述三氨基苯硼酸或其衍生物和硫脲在酸性混合溶液中的浓度分别独立为5~20mg/ml,10~15mg/ml,优选12~14mg/ml,例如5mg/ml、6mg/ml、7mg/ml、8mg/ml、9mg/ml、10mg/ml、11mg/ml、12mg/ml、13mg/ml、14mg/ml、15mg/ml、16mg/ml、17mg/ml、18mg/ml、19mg/ml、20mg/ml等。
20.在本发明的一些实例中,所述酸性混合溶液与水热反应采用的容器体积比为1:2~10,优选1:3~8,更有选1:4~6,例如1:2,1:3,1:4,1:5,1:6,1:7,1:8,1:9,1:10等。
21.在本发明的一些实例中,所述水热反应结束后还包括过滤的步骤。所述过滤可采用孔径为0.1~0.5μm的滤膜,优选孔径为0.2~0.3μm。
22.本发明第二方面是提供上述制备方法得到的硼硫掺杂碳量子点。
23.在本发明的一些实例中,所述硼硫掺杂碳量子点主要由b、s、c、n、o五种元素组成。
24.在本发明的一些实例中,所述硼硫掺杂碳量子点的粒径分布为0.5~10nm,优选1~8nm,更优选1.5~5.5nm。所述硼硫掺杂碳量子点的平均粒径为2~5nm,优选3~4nm。
25.本发明第三方面是提供所述硼硫掺杂碳量子点在制备比率荧光传感器中的应用。
26.本发明第四方面是提供所述硼硫掺杂碳量子点在检测恩诺沙星中的应用。
27.本发明第五方面是提供一种恩诺沙星的检测方法,包括如下步骤:将所述硼硫掺杂碳量子点和待测样品共孵育后进行荧光检测,根据荧光强度的变化来测得待测样品中恩诺沙星的浓度。
28.在本发明的一些实例中,所述荧光检测采用的激发波长为270~290nm,优选280~290nm,更优选285nm。在该激发波长下,硼硫掺杂碳量子点和恩诺沙星共孵育的混合物发出两种不同波长的荧光。
29.在本发明的一些实例中,根据荧光强度的变化来测得待测样品中恩诺沙星的浓度具体为:记录305~550nm波长范围内的荧光光谱变化,根据荧光光谱中两个荧光峰强度的比值与恩诺沙星浓度之间的关系获得待测样品中恩诺沙星的浓度。所述两个荧光峰的比值具体为455nm和331nm处的两个荧光峰强度的比值,更具体为f
455
/f
331
。
30.在本发明的一些实例中,所述共孵育的温度为0~50℃,优选10~40℃,更优选20~30℃。
31.在本发明的一些实例中,所述共孵育的时间为1~30min,优选5~25min,更优选10~15min。
32.相对于现有技术,本发明具有如下有益效果:
33.(1)本发明通过采用三氨基苯硼酸获取衍生物作为原料,可同时作为碳源和硼源,且三氨基苯硼酸具有苯环,有利于碳量子点共轭结构的产生,与硫脲进行反应后可制得特殊的硼硫掺杂碳量子点,该硼硫掺杂碳量子点粒径均匀且具有较好的荧光特性,能够对作为比率荧光传感器对抗生素恩诺沙星进行特异性检测。
34.(2)本方法合成步骤简单、操作方便,合成速度快,产物稳定;反应无任何有害气液体产生,符合绿色安全环保理念。
附图说明
35.图1为b,s-cds(硼硫掺杂碳量子点)的合成流程及对enr(恩诺沙星)的比率检测示意图;
36.图2为b,s-cds的透射电镜图(a)以及粒径分布直方图(b);
37.图3为b,s-cds的红外分析图;
38.图4为b,s-cds的元素分析图;
39.图5为不同体系的紫外吸收光谱图(a)和b,s-cds的荧光光谱图(b);
40.图6为b,s-cds与不同浓度的enr体系的荧光光谱图(a)和enr检测线性方程;
41.图7为b,s-cds特异选择性检测结果(a)和抗干扰性检测结果(b)。
具体实施方式
42.以下结合具体的实施例进一步说明本发明的技术方案。以下实施例中所用的原料,如无特殊说明,均可从常规商业途径得到;所采用的工艺,如无特殊说明,均采用本领域的常规工艺。
43.实施例1
44.本实施例合成了一种硼硫掺杂碳量子点(b,s-cds),并将其应用于对enr(恩诺沙星)的比率检测,合成流程及对enr的比率检测示意图如图1所示。
45.其中b,s-cds的制备方法具体包括如下步骤:
46.(1)分别称量0.0685g三氨基苯硼酸一水合物、0.038g的硫脲并将其溶解于5ml、0.3m的稀硝酸溶液中,超声5min后使其完全溶解;
47.(2)将上述溶液转移至25ml四氟乙烯反应釜中,置于180℃恒温烘箱中反应4h后自然冷却至室温;
48.(3)将步骤(2)所得材料进行0.22μm的有机相滤膜过滤以除去大颗粒杂质,即可得b,s-cds储备液。该b,s-cds储备液置于4℃下储存,每次使用时取20μl进行稀释即可。
49.如图2所示为b,s-cds的透射电镜图(a)以及粒径分布直方图(b)。从图中可以看出,合成的b,s-cds呈均匀的球状分布,且其具有较好的分散性。根据对b,s-cds进行粒径分布分析可知,其粒径范围在1.5~5.5nm之间,平均粒径为3.4nm,这也进一步说明b,s-cds成功合成。
50.图3为b,s-cds的红外分析图,可见b,s-cds保留了原料的较多双键结构(共轭结构),这将有利于提高b,s-cds的荧光性能。
51.图4为b,s-cds的元素分析图,可见合成的b,s-cds主要由b,s,c,n,o五种元素组成,这与合成原料的元素组成一致。
52.对enr的比率检测过程和结果如下:
53.(1)激发波长确定
54.取20μl的b,s-cds储备液与一定浓度的enr(恩诺沙星)储备液(溶剂为20%甲醇溶液)于5ml塑料管中,并用pbs缓冲溶液(ph=5.5)定容至4ml后(定容后enr的浓度为30μm)置于超声环境中孵育10min以保证溶液混合均匀。对共孵育后的体系进行紫外吸收测试和荧光测试。
55.作为比较,在相同条件下对单独的enr溶液(30μm)与单独的b,s-cds储备液进行荧光测试以及紫外吸光测试,结果如图5所示,图5(a)沿箭头方向的曲线依次为b,s-cds+enr,b,s-cds,enr。图5显示,b,s-cds在285nm处有一个明显的紫外吸收峰(a),结合荧光光谱图(b)最终确定体系最佳激发波长为285nm。
56.(2)enr检测
57.取20μl的b,s-cds储备液与系列不同浓度的enr储备液(溶剂为20%甲醇溶液)于5ml塑料管中,并用pbs缓冲溶液(ph=5.5)定容至4ml(定容后enr浓度为0~30μm)后置于超声环境中孵育10min以保证溶液混合均匀。在500v电压下,选择5nm的激发和发射狭缝宽度,在激发波长为285nm下记录不同体系305~550nm荧光光谱。
58.如图6(a)所示,不同enr浓度下的荧光光谱在331nm处的荧光强度随enr浓度增加逐渐减弱,而在455nm处出现一个新的荧光发射峰并随着目标分析物enr浓度增加而逐渐增强,说明b,s-cds可作为enr检测的比率荧光传感器,可以通过分析f
455
/f
331
比率荧光值来进行enr检测。以f
455
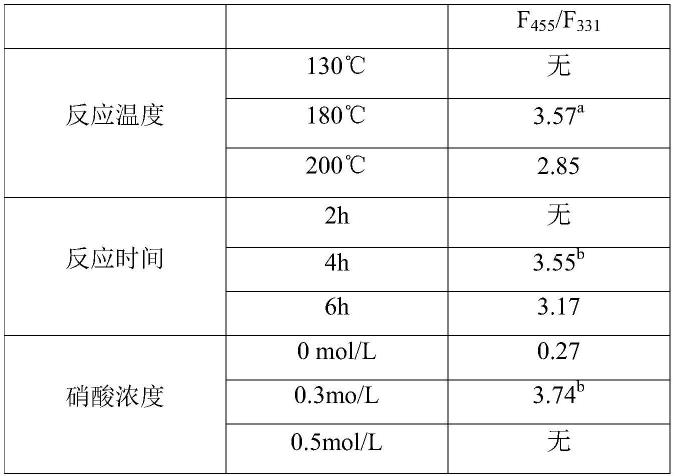
/f
331
比率荧光值为纵坐标,enr浓度为横坐标构建线性方程如图6(b)所示,该线性方程为y=0.1256x-0.1252,r2=0.9908。该检测体系的检测限为0.1158μm,灵敏度好。
59.(3)特异选择性
60.取20μl的b,s-cds储备液与一定浓度的enr储备液、加替沙星、巴洛沙星、克林沙星、莫西沙星、诺氟沙星、司帕沙星或左氧氟沙星(溶剂为20%甲醇溶液)于5ml塑料管中,并
用pbs缓冲溶液(ph=5.5)定容至4ml后(定容后各抗生素的浓度为30μm)置于超声环境中孵育10min以保证溶液混合均匀。在500v电压下,选择5nm的激发和发射狭缝宽度,在激发波长为285nm下记录不同体系305~550nm荧光光谱,并分析f
455
/f
331
比率荧光值。
61.不同检测体系中的f
455
/f
331
比率荧光值如图7(a)所示。结果显示,b,s-cds对enr具有最高的f
455
/f
331
比率荧光值,因此可b,s-cds可作为enr的比率荧光探针。而对其他的抗生素f
455
/f
331
比率荧光值≤1,无法通过比率值来进行测试。可见,b,s-cds对enr具有很好的特异性识别能力。
62.(4)抗干扰性
63.取20μl的b,s-cds储备液与一定浓度的enr储备液(溶剂为20%甲醇溶液)以及干扰离子(ag
+
、ba
2+
、ca
2+
、cu
2+
、k
+
、mg
2+
、mn
2+
、na
+
、pb
2+
、zn
2+
、cl-、co
32-、f-或no
3-)于5ml塑料管中,并用pbs缓冲溶液(ph=5.5)定容至4ml(定容后干扰离子的浓度为150μm)后置于超声环境中孵育10min以保证溶液混合均匀。在500v电压下,选择5nm的激发和发射狭缝宽度,在激发波长为285nm下记录不同体系305~550nm荧光光谱,并分析f
455
/f
331
比率荧光值。
64.同时,在b,s-cds储备液中加入相同量的干扰离子并稀释到相同的浓度,然后进行荧光测试。
65.不同干扰离子下b,s-cds体系以及b,s-cds和enr的混合体系的f
455
/f
331
比率荧光值如图7(b)所示。图中深色的表示b,s-cds+ions,即在b,s-cds中加入干扰离子的实验组;浅色的表示b,s-cds+ions+enr,即在b,s-cds和enr的混合体系中加入干扰离子的实验组。结果显示,加入不同干扰离子后,两种体系的f
455
/f
331
比率荧光值与空白组相比几乎没有发生变化,或者只发生少量变化,说明b,s-cds的抗干扰能力强。
66.实施例2
67.本实施例合成了一种b,s-cds,其制备方法与实施例1的唯一区别在于:b,s-cds的制备方法的步骤(2)中反应温度为130℃或200℃。
68.实施例3
69.本实施例合成了一种b,s-cds,其制备方法与实施例1的唯一区别在于:b,s-cds的制备方法的步骤(2)中反应时间为2h或6h。
70.实施例4
71.本实施例合成了一种b,s-cds,其制备方法与实施例1的唯一区别在于:b,s-cds的制备方法的步骤(1)中的硝酸浓度为0或者0.5m。
72.取20μl的实施例1~4的b,s-cds储备液与enr储备液于5ml塑料管中,并用pbs缓冲溶液(ph=5.5)定容至4ml(定容后enr浓度为30μm)后置于超声环境中孵育10min以保证溶液混合均匀。在500v电压下,选择5nm的激发和发射狭缝宽度,在激发波长为285nm下记录不同体系305~550nm荧光光谱,并分析f
455
/f
331
比率荧光值。
73.实施例1~4制得的b,s-cds在加入相同浓度enr后的f
455
/f
331
比率荧光值如下表1所示。
74.表1.不同b,s-cds+enr的f
455
/f
331
比率荧光值
[0075][0076]a、b、c:三者均为实施例1条件下制得的b,s-cds,三个数值在误差允许范围内。
[0077]
根据表1可知,制备b,s-cds过程中反应温度过低,或者反应时间过少,得到的产物无法对enr产生比率荧光;同时,在非酸性条件下得到的b,s-cds对enr的荧光比率过低,会影响检测,而酸度过高则会导致无比率荧光。
[0078]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1