一种Si-C新型有机硅季铵盐表面活性剂及制备方法
一种si-c新型有机硅季铵盐表面活性剂及制备方法
技术领域
1.本发明涉及季铵盐表面活性剂制备技术领域,特别涉及一种si-c新型有机硅季铵盐表面活性剂及制备方法。
背景技术:
2.有机硅表面活性剂是随着有机硅新型材料发展起来的一种新型表面活性剂,其疏水基团(si-o-si等)比传统的碳链疏水性更强,具有优良的降低表面张力的能力,是一类高效的表面活性剂,因此也成为表面活性剂领域的研究热点之一。
3.有机硅表面活性剂是由聚硅氧烷和亲水基组成,其结构通式如下式所示。其中当r为聚氧乙烯醚类非离子基团,则为非离子有机硅表面活性剂;r为磺酸基、羧基和磷酸基等极性基团,则为阴离子表面活性剂;r含有季铵盐、咪唑盐、吡咯盐、吡啶盐等结构单元时为阳离子有机硅表面活性剂甜菜碱型;r为氨基酸型和咪唑啉型等基团则为两性离子型有机硅表面活性剂。
[0004][0005]
目前,低表面张力的有机硅表面活性剂,几乎都是以三硅氧烷为疏水链,虽然cmc低,降低表面张力效果优异,但由于si原子体积大,易极化,又有3d空轨道可供成键,加上si-o键自身极性很大,故易与质子酸、无机酸酐、碱金属氢氧化物、水及醇等发生反应,会导致表面活性降低,使用时往往需要现配现用,在水体系中往往要在24小时用完,导致使用不便。另外,大多数关于有机硅表面活性剂的研究主要集中在非离子型的制备上,有关阳离子型有机硅表面活性剂的相关报道还很少,故开发一种高效、耐水解的si-c阳离子型有机硅表面活性剂十分必要。本发明直接通过季铵化反应,合成一系列含烯丙基的阳离子单体,再与三烷基硅烷,利用硅氢化反应,合成si-c阳离子型有机硅表面活性剂。
技术实现要素:
[0006]
本发明所要解决的技术问题:解决现有有机硅表面活性剂合成过程中存在的上述问题。
[0007]
为解决上述技术问题,本发明提供以下的技术方案:
[0008]
一种si-c有机硅季铵盐表面活性剂,季铵盐阳离子型有机硅表面活性剂的结构式如式(1)所示:
[0009][0010]
其中,其中,r为乙基、异丙基、己基、苯基中的一种或多种;n为10,14,16。
[0011]
优选地,所述r为乙基或异丙基或己基或苯基。
[0012]
上述si-c季铵盐阳离子型有机硅表面活性剂的合成过程如下:
[0013]
1)催化剂的制备:按四氢呋喃与氯铂酸的比例为20-50:1,将氯铂酸溶于无水四氢呋喃中,获得四氢呋喃与氯铂酸的络合物,该络合物参与反应的量为反应总量的1-1000ppm。四氢呋喃做无水干燥处理;所述干燥方法为:加入氢化钙重蒸处理。
[0014]
2)阳离子中间体的制备:反应容器中,按胺溴比1-1.3的摩尔比加入烯丙基二甲基胺与1-溴代直链烷,再按锌胺比0.02-0.08的摩尔比添加锌粉,维持50-80℃下反应10-18h。反应结束后减压旋蒸,乙醚萃取,45℃真空干燥5h制得烯丙基二甲基直链烷基季铵盐单体。制备阳离子中间体的化学反应式如下:
[0015][0016]
3)si-c新型有机硅季铵盐表面活性剂的制备:氮气保护下,将步骤1)和步骤2)中得到催化剂和阳离子中间体按一定的比例于80℃下搅拌1h,滴加一定量的烷基硅烷,反应制得。制备si-c有机硅季铵盐表面活性剂的化学反应式如下:
[0017][0018]
所述步骤3)中以三烷基硅烷为参照,干燥的四氢呋喃为溶剂,按三烷基硅烷与阳离子中间体的摩尔比例1.1-1.3,催化剂与三烷基硅烷的摩尔比例(pt:si-h)为100-1000ppm,在70-90℃下反应5-8h,减压旋蒸,45℃真空干燥5h得产品。
[0019]
优选地,所述1-溴代直链烷为1-溴代十二烷、1-溴代十六烷、1-溴代十八烷中的一种或多种。
[0020]
优选地,所述三烷基硅烷为三乙基硅烷、三异丙基硅烷、三己基硅烷、三苯基硅烷中的一种或多种。
[0021]
本发明获得的有益效果:
[0022]
1)本发明提供了一种si-c新型有机硅季铵盐表面活性剂,与传统表面活性剂相比具有高效性,且具有较低的临界胶束浓度。
[0023]
2)本发明提供了一种高效的新型有机硅季铵盐表面活性剂制备方法,该合成方法简单,条件温和,操作简便易于控制,后处理简单,产物易于分离提纯。
附图说明
[0024]
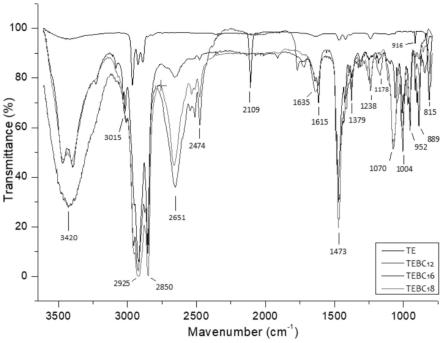
图1为三乙基硅烷(te),二甲基十二烷基三乙基硅基丙基溴化铵(tebc
12
),二甲基十六烷基三乙基硅基丙基溴化铵(tebc
16
),二甲基十八烷基三乙基硅基丙基溴化铵(tebc
18
)红外色谱图;
[0025]
图2中a~d依次为二甲基十二烷基三乙基硅基丙基溴化铵(tebc
12
),二甲基十二烷基三异丙基基硅基丙基溴化铵(tpbc
12
),二甲基十二烷基三己基硅基丙基溴化铵(thbc
12
),二甲基十二烷基三苯基硅基丙基溴化铵(tbbc
12
)核磁氢谱;
[0026]
图3中a~d依次为二甲基十二烷基三乙基硅基丙基溴化铵(tebc
12
),二甲基十二烷基三异丙基基硅基丙基溴化铵(tpbc
12
),二甲基十二烷基三己基硅基丙基溴化铵(thbc
12
),二甲基十二烷基三苯基硅基丙基溴化铵(tbbc
12
)核磁碳谱;
[0027]
图4为二甲基十八烷基三异丙基基硅基丙基溴化铵tpbc
18
水溶液的表面张力与浓度关系图。
[0028]
图5为二甲基十八烷基三异丙基基硅基丙基溴化铵tpbc
18
水溶液的电导率与浓度关系图。
具体实施方式
[0029]
下面通过对实施例的描述,对本发明的具体实施方式作进一步详细的说明,以帮助本领域的技术人员对本发明的发明构思、技术方案有更完整、准确和深入的理解。
[0030]
实施例1:按如下方法制备有机硅季铵盐表面活性剂:
[0031]
氯铂酸催化剂的制备:由0.9711g氯铂酸溶于10ml蒸馏除水处理的thf中制备得到,并在使用前保存在冰箱中。将1.0ml该溶液转移到100ml容量瓶中,用干燥的thf稀释至刻度线,混合后避光低温保存,可用于硅氢化反应。
[0032]
第一步:二甲基十二烷基烯丙基溴化铵(bc
12
)的制备:
[0033]
将配有温度计和回流冷凝管的三口烧瓶置于磁力搅拌器控温的水浴中,加40ml无水四氢呋喃、8.62g(0.1mol)n,n-二甲基丙烯胺、24.92g(0.11mol)1-溴代十二烷和0.33g(0.005mol)锌粉,在65℃下进行反应12h,反应完毕。于45℃下,减压旋蒸四氢呋喃,分别用30ml无水乙醚洗涤5次,45℃真空干燥5h得产品。本发明实施例中的无水四氢呋喃由四氢呋喃加氢化钙重蒸制得。
[0034]
第二步:二甲基十二烷基三乙基硅基丙基溴化铵(tebc
12
)的制备:
[0035]
将二甲基十二烷基烯丙基溴化铵(0.01mol)和4ml氯铂酸催化剂加入装有搅拌器、回流冷凝管和温度计的三颈瓶中,通氮气,80℃下恒温搅拌1h,再滴加三乙基硅烷(0.011mol),保温反应5h,之后,蒸出溶剂,再减压蒸馏脱除低沸物,冷却,得白色粘稠固体,45℃真空干燥5h即得产品tebc
12
。
[0036]
实施例2:按如下方法制备有机硅季铵盐表面活性剂:
[0037]
第一步:二甲基十二烷基烯丙基溴化铵(bc
16
)的制备:
[0038]
将配有温度计和回流冷凝管的三口烧瓶置于磁力搅拌器控温的水浴中,加40ml无水四氢呋喃、12.92g(0.15mol)n,n-二甲基丙烯胺、50.38g(0.165mol)1-溴代十六烷和0.49g(0.0075mol)锌粉,在65℃下进行反应10h,反应完毕。于45℃下旋蒸四氢呋喃,分别用30ml无水乙醚洗涤5次,45℃真空干燥5h得产品。
[0039]
第二步:二甲基十六烷基三异丙基硅基丙基溴化铵(tpbc
16
)的制备:
[0040]
将二甲基十六烷基烯丙基溴化铵(0.01mol)和2ml氯铂酸催化剂加入装有搅拌器、回流冷凝管和温度计的三颈瓶中,通氮气,80℃下恒温搅拌1h,再滴加三异丙基硅烷(0.012mol),保温反应8h,之后,蒸出溶剂,再减压蒸馏脱除低沸物,冷却,得白色固体,45℃
真空干燥5h即得产品tpbc
16
。氯铂酸催化剂的制备方法与实施例1中相同,不同之处在于四氢呋喃与氯铂酸的体积/质量比为20:1。
[0041]
实施例3:按如下方法制备有机硅季铵盐表面活性剂:
[0042]
第一步:二甲基十八烷基烯丙基溴化铵(bc
18
)的制备:
[0043]
将配有温度计和回流冷凝管的三口烧瓶置于磁力搅拌器控温的水浴中,加40ml无水四氢呋喃、8.62g(0.1mol)n,n-二甲基丙烯胺、24.92g(0.11mol)1-溴代十八烷和0.33g(0.005mol)锌粉,在50℃下进行反应18h,反应结束。于45℃下,旋蒸四氢呋喃,分别用30ml无水乙醚洗涤5次,45℃真空干燥5h得产品。
[0044]
第二步:二甲基十八烷基三己基硅基丙基溴化铵(tpbc
18
)的制备:
[0045]
将二甲基十八烷基烯丙基溴化铵(0.01mol)和6ml氯铂酸催化剂加入装有搅拌器、回流冷凝管和温度计的三颈瓶中,通氮气,80℃下恒温搅拌1h,再滴加三己基硅烷(0.013mol),保温反应6h,之后,蒸出溶剂,再减压蒸馏脱除低沸物,冷却,得白色固体,45℃真空干燥5h即得产品tpbc
18
。氯铂酸催化剂的制备方法与实施例1中相同,不同之处在于四氢呋喃与氯铂酸的体积/质量比为35:1。
[0046]
实施例4:按如下方法制备有机硅季铵盐表面活性剂:
[0047]
第一步:二甲基十八烷基烯丙基溴化铵(bc
18
)的制备:
[0048]
将配有温度计和回流冷凝管的三口烧瓶置于磁力搅拌器控温的水浴中,加40ml无水四氢呋喃、8.62g(0.1mol)n,n-二甲基丙烯胺、24.92g(0.11mol)1-溴代十八烷和0.33g(0.005mol)锌粉,在80℃下进行反应10h,反应结束。于45℃下,旋蒸除去溶剂四氢呋喃,分别用30ml无水乙醚洗涤5次,45℃真空干燥5h得产品。
[0049]
第二步:二甲基十八烷基三苯基硅基丙基溴化铵(tbbc
18
)的制备:
[0050]
将二甲基十八烷基烯丙基溴化铵(0.01mol)和8ml氯铂酸催化剂加入装有搅拌器、回流冷凝管和温度计的三颈瓶中,通氮气,80℃下恒温搅拌1h,再滴加三苯基硅烷(0.013mol),保温反应8h,之后,蒸出溶剂,再减压蒸馏脱除低沸物,冷却,得白色固体,45℃真空干燥5h即得产品tbbc
18
。氯铂酸催化剂的制备方法与实施例1中相同,不同之处在于四氢呋喃与氯铂酸的体积/质量比为50:1。
[0051]
如图1所示,朝向2925cm-1
和2850cm-1
的峰值归因于c-h拉伸振动,朝向1473cm-1
的峰值归因于c-h弯曲振动。由于长链烷基(c
12
,c
16
,c
18
)的引入,吸收带变得更宽更强。季铵基团c-n拉伸的吸收峰在1379cm-1
处观察,如图1所示。由于c-n的吸收带从1000cm-1
到1200cm-1
,1070cm-1
和1004cm-1
峰的吸收增强。1238cm-1
和815cm-1
处吸收峰分别表示si-c的弯曲振动和拉伸振动。在1178cm-1
处出现一特征峰,对应于c-h(si-ch
2-)弯曲振动,对于te的加氢硅化试剂,2109cm-1
和916cm-1
处的峰表示si-h的拉伸振动和弯曲振动。烯丙基氢化后两个峰消失,均表明已经合成了目标产物。
[0052]
如图2所示,首先,对于si-h基团,我们没有发现在4.9-5.1ppm处的峰,这表明氢甲硅烷基化反应完成。该观察结果与ir光谱一致。其次,有代表性的1h nmr峰可归属于c
12
长链中的甲基(0.8-0.9ppm,峰b),亚甲基(-(ch2)
n-)(1.2-1.4ppm,峰d),分别对应表面活性剂a和c,可以将在0.5-0.6ppm(峰a)处检测到的信号指定为亚甲基(si-ch
2-)。同时,对于表面活性剂a和b,可以将在0.9-1.1ppm(峰c)处检测到的信号指定为来自碳硅烷基团(si-ch
2-ch3和si-ch-(ch3)2)上的甲基。对于表面活性剂d,7.3-7.7ppm(峰m)处的信号对应于苯环的
质子。将1.7-1.9ppm处的峰(峰e)指定为亚甲基(n-c-ch
2-)。对于后两组,随着反应的进行,化学位移数据转移到更高的场,即峰g和峰f在所有情况下,ftir和1h nmr的光谱数据与化合物的指定结构一致。
[0053]
图3中以tebc
12
(a)为例:δ(ppm)=6.3(sich2ch3),6.7(sich2ch3),14.1(ch2ch2ch3),22.6(ch2ch2ch3),24.2(nch2ch2),26.6(sich2ch2),29.0-29.5((ch2)nch2ch3),31.8(ch2ch2ch3),50.6(n
+
ch3,a),57.9(n
+
ch3,bc
12
),63.9(n
+
ch2ch2ch2),66.1(sich2ch2ch2,a),68.6(ch2chch2,bc
12
),124.3(ch2chch2,bc
12
),129.8(ch2chch2,bc
12
).
[0054]
以上实施例仅为说明本发明的技术思想,不能以此限定本发明的保护范围,凡是按照本发明提出的技术思想,在技术方案基础上所做的任何改动,均落入本发明保护范围之内;本发明未涉及的技术均可通过现有技术加以实现。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1