一种可降解性压敏胶的制备方法
1.本发明涉及高分子材料及化学化工领域,具体地,本发明涉及一种可降解聚酯制备压敏胶的制备方法。
背景技术:
2.由于与石油产品的使用有关的环境和其他问题,可降解聚合物材料受到了极大的关注。目前,可降解聚合物被用于生物医学、食品包装和一次性物品,但其有限的性质限制了它们的广泛应用。压敏胶(psa)代表了一种潜在的应用(例如,胶带、标签、邮票和粘贴纸),这将进一步拓展可降解聚酯的应用范围。传统的psa材料大多是由聚丙烯酸酯类、橡胶类、聚氨酯类等构成。在回收过程中,这些压敏胶会被搅碎,产生的大粘性物质被有效去除,但仍留下微粘性物质导致回收行业的所谓“粘性”的问题。这些粘性物质会沉积在设备上,导致回收材料质量的下降和回收成本的增加。
3.解决压敏胶在回收过程中残留的“粘性物质”问题的一个方法就是制备可降解性压敏胶。可降解性压敏胶可以在比较温和的降解条件下或者添加绿色助剂的情况下可以降解成为小分子。目前对于可降解压敏胶的研究仍然较少,文献报道的这类材料有menthide与la的共聚物
1.、menthide与membl的共聚物
2.、pentadecyl caprolactone与la的共聚物
3.。这些聚合物除了拥有较强的粘附力之外,由于其主链中含有酯键,可以通过加热的方式使其断裂,从而实现它的可降解性。其中我们发现聚1,5-二恶烷-2-酮(pdxo)是与文献报道的压敏胶材料极其相似的一种聚醚酯,其单体1,5-二恶烷-2-酮(dxo)早在很久之前已经实现商品化生产,可以使用较为成熟的方法制备。
4.虽然dxo的制备方法比较成熟,但是目前大多数文献报道的dxo的开环聚合的催化剂仅仅围绕着辛酸亚锡、异丙醇铝、有机强碱等。使用这些催化剂面临着聚合能力差、聚合温度高、聚合时间长、分子量和分布均不可控等问题。近几年来,二元催化体系在环内酯的开环聚合中表现出极强的催化活性和控制性,这其中路易斯酸碱对体系得到了较为广泛的使用,它独特地利用了路易斯酸(la)和路易斯碱(lb)两种组分来协同催化聚合各种单体,通过平衡la和lb的酸碱性以及它们的空间相互作用,来显著影响链引发、链增长、链终止和链转移。
5.有鉴于此,本发明使用了有机碱和二乙基锌组成的路易斯酸碱对体系,实现dxo的快速可控开环聚合以制备可降解压敏胶材料。本发明提供的方法与以往报道的方法相比,具有以下有益之处:1)所使用的有机碱和二乙基锌易得、毒性低、并且易于从产物中除去;2)所用催化体系活性高,聚合时间短,并且能够在温和的条件下实现dxo可控的开环聚合,所得的聚酯产品能够做为很好的压敏胶使用。
技术实现要素:
6.本发明的目的是提供一种1,5-二恶烷-2-酮(dxo)快速可控开环聚合以制备可降解性压敏胶的方法,包括如下步骤:
7.(1)将引发剂、有机碱和二乙基锌溶于有机溶剂中,在一定温度下搅拌一段时间;
8.(2)将1,5-二恶烷-2-酮(dxo)加入上述混合溶液中,在一定温度下反应一段时间,加入酸性物质终止反应,将反应混合物加入甲醇中沉淀得到聚1,5-二恶烷-2-酮(pdxo)。
9.上述制备方法中,所述聚1,5-二恶烷-2-酮(pdxo)的化学结构式如式(ⅰ)所示:
[0010][0011]
上述的制备方法中,所述有机碱为其中一种,其中有机碱为式(ⅱ)、(iii)和(ⅳ)所示化合物。
[0012][0013]
上述制备方法中,步骤(1)中所述引发剂为醇,具体可为苄醇和邻苯二甲醇;所述有机碱可为1,8-二氮杂双环[5.4.0]十一碳-7-烯(dbu),4-二甲氨基吡啶(dmap),1,5,7-三氮杂二环[4.4.0]癸-5-烯。所述有机碱与二乙基锌的摩尔比例为0.5/1~3/1。
[0014]
上述制备方法中,步骤(1)中所述温度为室温;所述搅拌时间为5min至10min;所述有机溶剂可为甲苯、四氢呋喃、n,n-二甲基甲酰胺。
[0015]
上述制备方法中,步骤(2)中所述反应温度为25-100℃;所述反应时间为2~16h,所述1,5-二恶烷-2-酮在体系中的摩尔浓度为3~8mol/l,所述单体与引发剂的摩尔比例为1000/1~50/1。
[0016]
上述制备方法中,所述酸性物质为乙酸、苯甲酸、盐酸,所述酸性物质与有机碱的摩尔比例为1/1~10/1。
附图说明
[0017]
图1为对比实施例1中1,5-二恶烷-2-酮的1h nmr谱图。
[0018]
图2为对比实施例1中制得的聚1,5-二恶烷-2-酮的1h nmr谱图。
[0019]
图3为实施例1至实施例5制得的聚1,5-二恶烷-2-酮的gpc谱图。
[0020]
图4为实施例1至实施例5制得的聚1,5-二恶烷-2-酮在扫描速率为10℃/min时的dsc
[0021]
谱图。
[0022]
图5为实施例5制得的聚1,5-二恶烷-2-酮压敏胶的剪切性能测试图。
[0023]
图6为实施例5制得的聚1,5-二恶烷-2-酮压敏胶的剥离强度测试图。
具体实施方式
[0024]
下述实施案例对本发明进行具体描述,但本发明不限于这些实施案例。
[0025]
下述实施案例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0026]
对比实施例1
[0027]
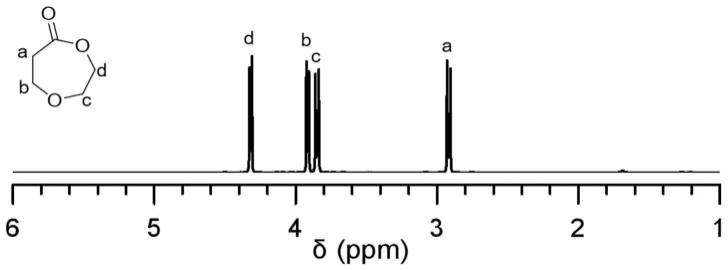
将(0.0384mmol,4.4mg)苄醇,(0.33mmol,40.8mg)二乙基锌溶于1.2ml甲苯中,置于室温中搅拌5min,将(3.45mmol,0.4g)1,5-二恶烷-2-酮加入反应管中。反应在氮气保护下进行16h,加入2滴醋酸终止反应。将反应混合物倒入10ml甲醇中,离心分离沉淀得到聚合物,即聚1,5-二恶烷-2-酮,其核磁氢谱如图2所示。
[0028]
实施例1
[0029]
(0.0096mmol,1.1mg)苄醇,(0.33mmol,40.8mg)二乙基锌溶于5ml甲苯中,置于50℃中搅拌5min,将(3.45mmol,0.4g)1,5-二恶烷-2-酮加入反应管中。反应在氮气保护下进行3h,加入2滴醋酸终止反应。将反应混合物倒入10ml甲醇中,离心分离沉淀得到聚合物,gpc测得数均分子量为15.7kg/mol,分子量分布为1.54,gpc谱图如图3所示。与对比实施例1相比较,增加反应温度,可加快聚合速度。
[0030]
实施例2
[0031]
(0.0096mmol,1.1mg)苄醇,(0.33mmol,40.8mg)二乙基锌,(0.33mmol,45.8mg)tbd溶于5ml甲苯中,置于50℃中搅拌5min,将(3.45mmol,0.4g)1,5-二恶烷-2-酮加入反应管中。反应在氮气保护下进行6h,加入2滴醋酸终止反应。将反应混合物倒入10ml甲醇中,离心分离沉淀得到聚合物,gpc测得数均分子量为21.7kg/mol,分子量分布为1.37,gpc谱图如图3所示,dsc谱图如图4所示。与对比实施例1相比较,增加反应聚合度,分子量呈现可控性增长。
[0032]
实施例3
[0033]
(0.0096mmol,1.1mg)苄醇,(0.33mmol,40.8mg)二乙基锌,(0.0165mmol,2mg)dmap溶于1.2ml甲苯中,置于室温中搅拌5min,将(3.45mmol,0.4g)1,5-二恶烷-2-酮加入反应管中。反应在氮气保护下进行6h,加入2滴醋酸终止反应。将反应混合物倒入10ml甲醇中,离心分离沉淀得到聚合物,gpc测得数均分子量为38.2kg/mol,分子量分布为1.60,gpc谱图如图3所示,dsc谱图如图4所示。与对比实施例1相比较,使用dmap/znet2显著缩短聚合时间,分子量呈现可控性增长。
[0034]
实施例4
[0035]
(0.0096mmol,1.1mg)苄醇,(0.33mmol,40.8mg)二乙基锌,(0.33mmol,5.5mg)dbu溶于1.2ml甲苯中,置于室温中搅拌5min,将(3.45mmol,0.4g)1,5-二恶烷-2-酮加入反应管中。反应在氮气保护下进行6h,加入2滴醋酸终止反应。将反应混合物倒入10ml甲醇中,离心分离沉淀得到聚合物,gpc测得数均分子量为20.6kg/mol,分子量分布为1.67。gpc谱图如图3所示,dsc谱图如图4所示。与对比实施例1相比较,使用dbu/znet2显著缩短聚合时间,分子量呈现可控性增长。
[0036]
实施例5
[0037]
(0.0035mmol,0.48mg)邻苯二甲醇,(0.33mmol,40.8mg)二乙基锌,(0.0165mmol,2mg)dmap溶于1.2ml甲苯中,置于室温中搅拌5min,将(3.45mmol,0.4g)1,5-二恶烷-2-酮加入反应管中。反应在氮气保护下进行6h,加入2滴醋酸终止反应。将反应混合物倒入10ml甲醇中,离心分离沉淀得到聚合物,gpc测得数均分子量为81.4kg/mol,分子量分布为1.38,gpc谱图如图3所示,dsc谱图如图4所示。与对比实施例1相比较,使用dmap/znet2可以制备较高分子量的聚1,5-二恶烷-2-酮。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1