丙烷选择氧化制丙烯醛和烯烃用催化剂及其制备与应用的制作方法
本发明涉及一种丙烷选择氧化制丙烯醛和烯烃用催化剂及其制备与应用,属于石油化工催化技术领域。
背景技术:
随着石油价格不断攀升,石油油品的不断恶化,以石油为原料的化工行业面临着诸多挑战。全球存在着丰富的低碳烷烃,特别是将丙烷转化更有价值的化工原料有着巨大的经济利益,通过选择性氧化过程完成这个转换引起人们的广泛关注。
丙烷分子由于具有较强的稳定性,化学反应活性很低。目前除少部分用于生产丙烯腈、裂解制乙烯等外,大部分丙烷用于燃烧,造成资源浪费。在丙烷选择氧化过程中,目的产物比反应物本身更容易深度氧化,故丙烷的高活性与其选择氧化产物的高选择性难以兼得。在提高丙烷活性的同时保持选择氧化产物分子稳定是丙烷直接氧化的关键和难点所在。在丙烷选择氧化反应中,生成醛类含氧化合物是很困难的催化过程,能够生成醛类含氧化合物的催化剂种类并不多,产物分布及其选择性随催化剂不同有所差异,反应条件对同一种催化剂的活性和选择性也有影响。用于丙烷选择氧化生成醛类含氧化合物的催化剂,主要包括杂多酸类催化剂、体相的金属氧化物催化剂和负载金属氧化物催化剂等。
杂多酸类催化剂研究现状:目前在丙烷一步氧化制丙烯酸反应中研究较多的为Keggin型杂多酸,丙烯酸的收率一般小于15%。影响Keggin型杂多酸催化性能的首要因素是其化学组成,通过不同的元素组合可对其理化性质进行系统的调变。目前报道较多的是部分取代的十二磷铝杂多酸,可能是其酸性、氧化性较为适中。总的来讲,杂多酸催化剂主要的缺点是比表面积小和热稳定性差,催化作用较差。
体相金属氧化物研究现状:丙烷选择氧化涉及多电子转移及催化剂的多方面性质,复合金属氧化物能够提供多功能多中心活性位,用于丙烷选择氧化的复合金属氧化物(MMO)催化剂主要是过渡金属氧化物催化剂,大多数含有Mo、V和Te等元素。Shishido等(T.Shishido,T.Konishi and I.Matsuura,et al.,Catal.Today,71(2001)77. 考察了MoVSbO催化剂的丙烷氧化制丙烯醛反应性能,反应温度为430℃时,丙烷转化率为6.8%,丙烯醛选择性为20.8%。催化剂由VSbO4,α-Sb2O4和高分散的钼氧物种Mo8O23、Mo9O26、Mo18O52等物相组成。丙烷在VSbO4上氧化脱氢生成丙烯,然后丙烯在高分散的钼氧物种上转化为丙烯醛。何益明等(何益明,伊晓东,黄传敬等.MoVBiTeO/SiO2对丙烷选择氧化制丙烯醛反应的催化性能.催化学报.2008,29(4):409-414)考察了MoVBiTeO/SiO2对丙烷选择氧化制丙烯醛反应的催化性能,其用浸渍法制备了一系列不同Mo/V比的MoVBiTeO/SiO2催化剂,考察了催化剂对丙烷选择氧化制丙烯醛反应的催化性能,结果表明,在MoBiTeO/SiO2催化剂中添加V组分时,催化剂的低温可还原性和催化活性明显提高。由于V组分具有较强的氧化性,Mo与V组分之间存在较强的相互作用,调变了催化剂的结构,并形成了氧化还原循环促进了催化剂中电子和O物种的传递,使催化剂的低温可还原性增强,催化活性提高。Mo组分有利于形成L酸位,而V组分有利于形成B酸位。当Mo/V摩尔比为6时,催化剂具有最高的丙烯醛收率(9.7%),此时丙烷的转化率为33.9%。
负载型金属氧化物催化剂研究现状:负载型催化剂主要体现载体对催化剂反应性能的影响,如分离活性中心,调变催化剂的酸碱性和氧化还原性等。要在担载型催化剂上获得较高的丙烯醛和丙烯酸选择性以及收率就要协调好催化剂中“脱氢”和“插氧”中心的作用。通过调变载体的性质和改善催化剂的制备方法,可以使催化剂表面的“脱氢”中心和“插氧”中心之间的作用更加协调,并建立合适的表面酸-碱平衡和使催化剂具有合适的氧化还原性,这样可以提高丙烯醛和丙烯酸的选择性和收率。
易晓东等(X.Yi,X.Sun,X.Zhang,et al.Highly dispersed MoVTeNbO/SiO2catalysts prepared by the sol-gel method for selective oxidation of propane to acrolein.Catalysis Communications.2009,10(12):1591-1594)报道了用溶胶-凝胶法制备高分散MoV0.3Te0.23Nb0.1Ox/SiO2催化剂并考察了其在丙烷选择性氧化制丙烯醛反应性能。反应的最好的结果是在催化剂(Mo+V+Te+Nb)/Si=2mol%时,丙烷的转化率为39.9%,丙烯醛的选择性达到45.9%,而丙烯和COx的选择性分别为14.0%和31.5%。和浸渍法相比,由溶胶-凝胶方法制备的催化剂的性能更高,这可能是因为溶胶凝胶方法制备的催化剂有更高的特定的表面积,更好的分散在载体上的活性位,以及更低的反应温度。这有助于催化剂氧化还原性能和抑制丙烷和产物的深度氧化。
综合上述的报道我们发现丙烷选择氧化制丙烯醛等有机化工原料在产物选择性方面取得了一些进展,但丙烷的转化活性和转化率仍然很低,虽然在有的催化剂上产物的选择性比较高,但是转化率过低导致产物的收率很低,不具备实际的生产意义。
因此,寻找一种高效的丙烷选择氧化制备丙烯醛和烯烃用催化剂,提高丙烷的转化活性,是本领域亟待解决的问题之一。
技术实现要素:
本发明的目的在于提供一种丙烷选择氧化制丙烯醛和烯烃用催化剂。
本发明的目的还在于提供一种上述丙烷选择氧化制丙烯醛和烯烃用催化剂的制备方法。
本发明的目的还在于提供上述丙烷选择氧化制丙烯醛和烯烃用催化剂在丙烷选择氧化制丙烯醛和烯烃中的应用。
为达上述目的,本发明提供了一种丙烷选择氧化制丙烯醛和烯烃用催化剂,其是以纯硅KIT-6介孔分子筛为载体、以钼的氧化物为活性组分、以钾的氧化物为助剂;所述钼的氧化物掺杂进入纯硅KIT-6介孔分子筛的骨架中;所述钾的氧化物以高分散的形式负载于所述纯硅KIT-6介孔分子筛的表面,并且,所述K、Mo、Si的原料摩尔比为0.05-0.5:0.1-8:100。
根据本发明所述的催化剂,优选地,所述钼的氧化物掺杂进入纯硅KIT-6介孔分子筛的骨架中是以三嵌段共聚物P123为模板剂,以盐酸、钼源(例如钼酸盐)、硅源(例如正硅酸乙酯,TEOS)、正丁醇为原料采用直接水热合成法实现的,制备得到的纯硅KIT-6介孔分子筛为骨架中含有钼的纯硅KIT-6介孔分子筛,记为Mo-KIT-6;
更优选所述钼源包括钼酸盐。
上述骨架掺杂一般是在分子筛合成过程中通过直接水热合成将活性组分直接固定在分子筛骨架的晶格中,不但分散了活性位,而且提高了活性位的稳定性。
根据本发明所述的催化剂,优选地,在所述直接水热合成法中,所述钼酸盐应先溶于蒸馏水中,然后再与三嵌段共聚物P123模板剂的溶液混合。
根据本发明所述的催化剂,优选地,所述钾的氧化物以高分散的形式负载于所述纯硅KIT-6介孔分子筛的表面是以钾盐和骨架中含有钼的纯硅KIT-6介孔分子筛为原料通过等体积浸渍、超声分散处理、真空干燥处理、空气干燥及焙烧处理实现的;
更优选所述钾盐包括硝酸钾。
上述高分散是指活性金属或助剂的一种分散状态,通常是在含量很低时存在(一般是指含量在1%以下时),如果含量过高则会出现金属的晶相聚集,这不利于反应的进行,这个概念目前在中外文献中都有很多引用,是比较常见的一个概念。
本发明还提供了上述丙烷选择氧化制丙烯醛和烯烃用催化剂的制备方法,其包括以下步骤:
制备模板剂溶液:将三嵌段共聚物P123模板剂溶于稀盐酸中,在恒温水浴中搅拌至三嵌段共聚物P123模板剂完全溶解,得到模板剂溶液;
配制钼源溶液:将钼酸盐固体溶于蒸馏水中,搅拌至钼酸盐完全溶解,得到钼源溶液;
混合:将钼源溶液缓慢加入模板剂溶液中,其中,钼源溶液与模板剂溶液的重量比为0.3-0.43:1,在水浴中搅拌混合,加入正丁醇,继续搅拌,得到均相溶液,然后缓慢滴入正硅酸乙酯,最后加快搅拌器转速使溶液剧烈搅拌,得到混合溶液;
晶化:将混合溶液进行晶化反应,然后经冷却、离心、抽滤、洗涤、干燥、焙烧处理,得到骨架中含有钼的纯硅KIT-6介孔分子筛;
负载钾:将钾盐溶于去离子水配制成钾盐溶液,将该溶液缓慢滴入骨架中含有钼的纯硅KIT-6介孔分子筛,迅速搅拌均匀后,进行超声分散处理,然后经真空干燥、空气干燥及焙烧处理,得到丙烷选择氧化制丙烯醛和烯烃用催化剂,记为K/Mo-KIT-6;
其中,所述钾盐、钼酸盐与正硅酸乙酯的添加量以所述K、Mo与Si的原料摩尔比换算得到。
根据本发明所述的制备方法,优选地,所述制备模板剂溶液的步骤为:将1-3重量份的三嵌段共聚物P123模板剂溶于17-21重量份的稀盐酸中,在30-40℃的恒温水浴中进行搅拌使三嵌段共聚物P123模板剂完全溶解,得到模板剂溶液;
更优选地所述磁力搅拌的时间为4-6h;
更优选所述稀盐酸的摩尔浓度为1.0-3.0mol/L;
更优选地,所述三嵌段共聚物P123模板剂的添加量为2重量份,所述稀盐酸的添加量为19重量份,所述恒温水浴的温度为35℃,所述磁力搅拌的搅拌时间为4h。
本发明的丙烷选择氧化制丙烯醛和烯烃用催化剂的制备方法中对三嵌段共聚物P123模板剂没有特殊要求,所有市售的三嵌段共聚物P123模板剂均可以应用于本发 明,并且在将其应用于丙烷选择氧化制丙烯醛和烯烃用催化剂的制备过程中,不需要对其进行纯化过程;在本发明的优选实施例中,使用的三嵌段共聚物P123模板剂为美国Sigma公司生产的平均分子量为5800的三嵌段共聚物P123模板剂。
根据本发明所述的制备方法,优选地,所述配制钼源溶液的步骤为:将0.006-0.6重量份的钼酸铵(其分子式为(NH4)6Mo7O24·4H2O)固体溶于55-59重量份的去离子水中,在30-40℃的恒温水浴中磁力搅拌3-5h使其形成均一溶液,得到钼源溶液;
更优选地,所述去离子水为57重量份,所述恒温水浴的温度为35℃。
根据本发明所述的制备方法,优选地,所述混合步骤为:将模板剂溶液与钼源溶液在30-40℃的恒温水浴中搅拌混合1-4h,加入1.9-3.3重量份的正丁醇,加入正丁醇之后继续搅拌1-2h,再加入4.2-10.3重量份的正硅酸乙酯,加入正硅酸乙酯之后继续搅拌24-48h,得到混合溶液;
更优选地,所述恒温水浴的温度为35℃,所述正丁醇的添加量为2.52重量份,所述加入正丁醇之后继续搅拌的时间为1h,所述正硅酸乙酯的添加量为6.42重量份,所述加入正硅酸乙酯之后继续搅拌的时间为24h。
根据本发明所述的制备方法,优选地,所述晶化步骤为:将所述混合溶液加入高压晶化反应釜中,在80-120℃下,晶化反应24-48h,然后经冷却至室温,再以3000-5000转/min的转速离心10-20min,抽滤,洗涤,最后经100-150℃干燥6-12h,530-580℃焙烧4-8h,得到所述骨架中含有钼的纯硅KIT-6介孔分子筛;
更优选所述洗涤为使用去离子水将滤液冲洗至无泡沫;
更优选所述焙烧的升温速率控制为1-2℃/min;
更优选地,所述晶化的温度为100℃;所述干燥的温度为100℃,干燥的时间为6h,所述焙烧的温度为550℃,焙烧的时间为6h。
根据本发明所述的制备方法,骨架中含有钼的纯硅KIT-6介孔分子筛的制备过程是将三嵌段共聚物P123模板剂溶在盐酸中,再将钼酸铵溶于原本溶三嵌段共聚物P123模板剂所需要的水量中,待三嵌段共聚物P123模板剂、钼酸铵均溶解后再将二者混合,这样既保证了二者能够充分溶解,同时也没有改变KIT-6合成原料的配比,此外,钼的加入非常容易引起部分KIT-6合成中的溶胶凝胶发生团聚,形成团聚物,这样部分Mo也相应的形成聚合物,这是我们不想看到的,因此在本发明中使用离心的方法将上述团聚物分离,以高效地将Mo掺杂进KIT-6介孔分子筛的骨架,进而得 到高浓度的、高分散的隔离活性位,提高Mo掺杂催化剂的质量。
根据本发明所述的制备方法,优选地,所述负载钾的步骤为:
将所需质量的硝酸钾溶于去离子水配制成硝酸钾溶液,将该溶液缓慢滴入骨架中含有钼的纯硅KIT-6介孔分子筛中,迅速搅拌均匀后,超声分散处理20-40min,得到经超声分散处理的样品;
更优选所述硝酸钾溶液与骨架中含有钼的纯硅KIT-6介孔分子筛的质量比为1.1:1;
将所述经超声分散处理的样品置于真空干燥箱中,常温下真空干燥2-6h,得到经真空干燥的样品;
更优选所述真空干燥箱的真空度为0.01MPa以下;
将所述经真空干燥的样品置于烘箱中,在100-150℃下干燥6-12h,得到经干燥的样品;
将所述经干燥的样品在530-580℃焙烧3-6h,得到丙烷选择氧化制丙烯醛和烯烃用催化剂;
更优选所述焙烧的升温速率控制为1-2℃/min;
更优选地,所述超声分散处理的时间为30min,所述真空干燥的时间为4h,所述干燥的温度为100℃,干燥的时间为8h,所述焙烧的温度为550℃,焙烧的时间为4h。
在本发明所述的丙烷选择氧化制丙烯醛和烯烃用催化剂的制备方法中所涉及的“重量份”均是采用同一重量标准进行定量的。
本发明所述的丙烷选择氧化制丙烯醛和烯烃用催化剂的制备方法包括以下步骤:
制备模板剂溶液:将1-3重量份的三嵌段共聚物P123模板剂溶于17-21重量份摩尔浓度为1.0-3.0mol/L的稀盐酸中,在30-40℃的恒温水浴中进行搅拌4-6h使三嵌段共聚物P123模板剂完全溶解,得到模板剂溶液;
配制钼源溶液:将所需质量的钼酸铵(其分子式为(NH4)6Mo7O24·4H2O)固体溶于55-59重量份的去离子水中,在30-40℃的恒温水浴中磁力搅拌3-5h使其形成均一溶液,得到钼源溶液;
混合:将模板剂溶液与钼源溶液在30-40℃的恒温水浴中搅拌混合1-4h,加入1.9-3.3重量份的正丁醇,加入正丁醇之后继续搅拌1-2h,再加入4.2-10.3重量份的正硅酸乙酯,加入正硅酸乙酯之后继续搅拌24-48h,得到混合溶液;
晶化:将所述混合溶液加入高压晶化反应釜中,在80-120℃下,晶化反应24-48h,然后经冷却至室温,再以3000-5000转/min的转速离心10-20min,抽滤,使用去离子水将滤液冲洗至无泡沫,最后经100-150℃干燥6-12h、530-580℃焙烧4-8h,得到所述骨架中含有钼的纯硅KIT-6介孔分子筛;所述焙烧的升温速率控制为1-2℃/min;
负载钾:将所需质量的硝酸钾溶于去离子水配制成硝酸钾溶液,将该溶液缓慢滴入骨架中含有钼的纯硅KIT-6介孔分子筛中,迅速搅拌均匀后,超声分散处理20-40min,得到经超声分散处理的样品,所述硝酸钾溶液与骨架中含有钼的纯硅KIT-6介孔分子筛的质量比为1.1:1;
将所述经超声分散处理的样品置于真空度为0.01MPa以下的真空干燥箱中,常温下真空干燥2-6h,得到经真空干燥的样品;
将所述经真空干燥的样品置于烘箱中,在100-150℃下干燥6-12h,得到经干燥的样品;
将所述经干燥的样品在530-580℃焙烧3-6h,得到丙烷选择氧化制丙烯醛和烯烃用催化剂;所述焙烧的升温速率控制为1-2℃/min;
其中,所述钾盐、钼酸盐与正硅酸乙酯的添加量以所述K、Mo与Si的原料摩尔比换算得到。
根据本发明所述的制备方法,在负载K时,将所述经超声分散处理的样品置于真空干燥箱中进行真空干燥处理,经真空干燥处理后可以使得K的分散度更高。
本发明还提供了上述丙烷选择氧化制丙烯醛和烯烃用催化剂在丙烷选择氧化制丙烯醛和烯烃中的应用。
本发明得到的骨架中含有钼的纯硅KIT-6介孔分子筛的有序度更高,更有利于反应物和产物的传输;同时,本发明的丙烷选择氧化制丙烯醛和烯烃用催化剂中Mo的分散度更高,活性位的浓度更大,更有利于催化活性的提高。
本发明提供的丙烷选择氧化制丙烯醛和烯烃用催化剂以具有三维螺旋交叉孔道结构的纯硅KIT-6介孔分子筛为载体,三维螺旋交叉孔道结构有利于活性组分的分散及物料的传输,上述纯硅KIT-6介孔分子筛为载体与SiO2、SBA-15以及MCM-48等载体相比,所得到的催化剂用于丙烷选择氧化制备丙烯醛和烯烃时具有较高的丙烷选择氧化活性;同时,本发明提供的上述催化剂以过渡金属钼的氧化物为活性组分并将其掺杂进纯硅KIT-6介孔分子筛的骨架中,将过渡金属钼的氧化物活性组分掺杂进载 体纯硅KIT-6介孔分子筛骨架中可以促进活性位的分散度和稳定性,在将该催化剂用于丙烷选择氧化制备丙烯醛和烯烃中,可进一步提高丙烷反应活性;本发明的催化剂以碱金属钾的氧化物作为助剂进行修饰,助剂碱金属钾的氧化物以高分散形式负载在载体表面可以调变催化剂的结构,在将该催化剂用于丙烷选择氧化制备丙烯醛和烯烃中,该催化剂的丙烷选择氧化活性能够得到更进一步地提高。
将本发明提供的丙烷选择氧化制丙烯醛和烯烃用催化剂应用于丙烷选择氧化制丙烯醛和烯烃的反应中,在本发明的优选实施例中,丙烷选择氧化反应的产物丙烯醛和烯烃的摩尔收率分别可以达到26.0%和27.5%,由于活性金属钼的高度分散和特殊的局部化学环境,导致催化剂在丙烷选择氧化反应中具有优异的反应性能。骨架掺杂的催化剂反应效果要明显好于浸渍法制备的催化剂(相同实验条件下,丙烯醛最好单程收率为9.2%),同时也远高于目前报道的丙烷选择氧化制丙烯醛催化剂的活性。
附图说明
图1为不同Mo、Si摩尔比条件下合成的Mo-KIT-6的XRD谱图,其中,谱线上的数值代表Mo、Si摩尔比;
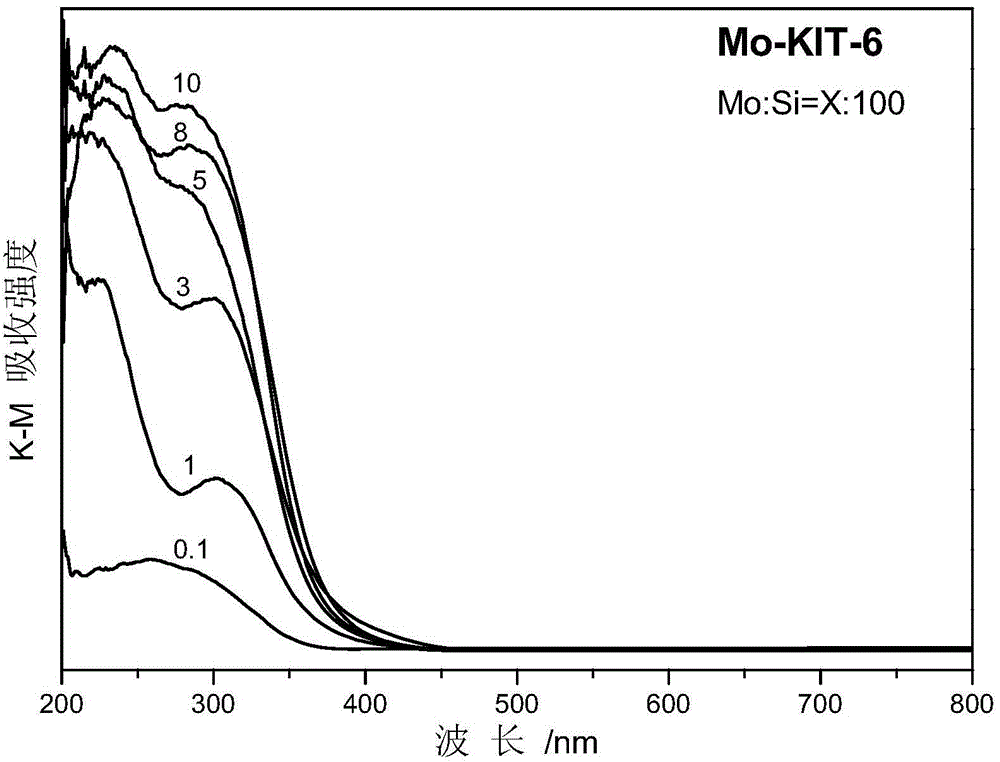
图2为不同Mo、Si摩尔比条件下合成的Mo-KIT-6的紫外可见漫反射谱图;
图3为实施例2中得到的丙烷选择氧化制丙烯醛和烯烃用催化剂(K/Mo-KIT-6催化剂)的UV-Vis谱图;
图4为实施例3中得到的丙烷选择氧化制丙烯醛和烯烃用催化剂(K/Mo-KIT-6催化剂)的UV-Vis谱图;
图5为实施例3中得到的丙烷选择氧化制丙烯醛和烯烃用催化剂(K/Mo-KIT-6催化剂)的XRD图;
图6为实施例1中Mo、Si摩尔比为8:100的条件下合成的Mo-KIT-6的透射电镜图;
图7为K、Mo、Si的摩尔比为0.1:8:100的K/Mo-KIT-6催化剂的透射电镜图;
图8为实施例1提供的Mo-KIT-6分子筛(Mo、Si摩尔比为0.1:100、1:100、8:100)和实施例2提供的K/Mo-KIT-6(K、Mo、Si摩尔比为0.1:0.1:100、0.1:1:100、0.1:8:100)催化剂在催化丙烷选择氧化制丙烯醛和烯烃的反应中丙烷转化率结果图;
图9为实施例1提供的Mo-KIT-6分子筛(Mo、Si摩尔比为0.1:100、1:100、8:100)和实施例2提供的K/Mo-KIT-6(K、Mo、Si摩尔比为0.1:0.1:100、0.1:1:100、0.1:8:100) 催化剂在催化丙烷选择氧化制丙烯醛和烯烃的反应中烯烃选择性的结果图;
图10为实施例1提供的Mo-KIT-6分子筛(Mo、Si摩尔比为0.1:100、1:100、8:100)和实施例2提供的K/Mo-KIT-6(K、Mo、Si摩尔比为0.1:0.1:100、0.1:1:100、0.1:8:100)催化剂在催化丙烷选择氧化制丙烯醛和烯烃的反应中丙烯醛选择性的结果图;
图11为实施例1提供的Mo-KIT-6分子筛(Mo、Si摩尔比为0.1:100、1:100、8:100)和实施例2提供的K/Mo-KIT-6(K、Mo、Si摩尔比为0.1:0.1:100、0.1:1:100、0.1:8:100)催化剂在催化丙烷选择氧化制丙烯醛和烯烃的反应中丙烯醛的收率结果图;
图12为实施例3提供的K/Mo-KIT-6催化剂在催化丙烷选择氧化制丙烯醛和烯烃的反应中丙烷转化率结果图;
图13为实施例3提供的K/Mo-KIT-6催化剂在催化丙烷选择氧化制丙烯醛和烯烃的反应中烯烃选择性结果图;
图14为实施例3提供的K/Mo-KIT-6催化剂在催化丙烷选择氧化制丙烯醛和烯烃的反应中丙烯醛选择性结果图;
图15为实施例3提供的K/Mo-KIT-6催化剂在催化丙烷选择氧化制丙烯醛和烯烃的反应中丙烯醛收率结果图;
图16为实施例1提供的Mo-KIT-6催化剂在催化丙烷选择氧化制丙烯醛和烯烃的反应中,接触时间与反应活性的结果图。
具体实施方式
以下将通过具体的实施例及附图详细地说明本发明的实施过程和产生的有益效果,旨在帮助阅读者更好地理解本发明的实质和特点,但是不作为对本案可实施范围的限定。
实施例1
本实施例提供了六种不同Mo、Si摩尔比条件下合成的Mo-KIT-6,其是按照以下步骤制备得到的:
将2g的三嵌段共聚物P123模板剂溶于19g摩尔浓度为2.0mol/L的盐酸中,在35℃的恒温水浴中磁力搅拌4h,使三嵌段共聚物P123模板剂完全溶解,形成均相溶液,得到三嵌段共聚物P123模板剂溶液;采用相同的操作条件制备得到六份均相三 嵌段共聚物P123模板剂溶液;
称取0.006g、0.055g、0.163g、0.272g、0.435g和0.544g的钼酸铵,分别将其与57g蒸馏水混合,在35℃的恒温水浴中磁力搅拌,使形成均一溶液,得到六份钼酸铵溶液。
将六份均相三嵌段共聚物P123模板剂溶液分别与相应的六份均相钼酸铵溶液混合后于35℃恒温水浴中搅拌2h,然后缓慢滴入2.52g正丁醇,继续搅拌1h后,逐滴加入6.42g正硅酸四乙酯,然后持续搅拌24h,得到六份混合溶液;
上述钼酸铵的质量是以Mo、Si摩尔比为0.1:100、1:100、3:100、5:100、8:100、10:100换算得到的。
将上述得到的六份混合溶液分别加入六个高压晶化反应釜中,在100℃下进行24h的晶化处理,晶化处理之后冷却至室温、再以4000转/min的转速离心15min,抽滤、使用蒸馏水将滤液冲洗至无泡沫,最后经过100℃干燥6h,550℃焙烧6h,所述焙烧的升温速率为2℃/min,得到六种骨架中含有钼的纯硅KIT-6介孔分子筛,即Mo-KIT-6。
上述六种Mo-KIT-6为Mo、Si摩尔比分别为0.1:100、1:100、3:100、5:100、8:100、10:100的六种Mo-KIT-6。
利用X射线粉末衍射对上述六种Mo-KIT-6分子筛进行检测,区分结晶相及非结晶相,表征分子筛材料的结晶度。根据不同位置的衍射峰及衍射峰的强度可以判断介孔分子筛材料形成与否及其有序度。
图1为不同Mo、Si摩尔比条件下合成的Mo-KIT-6的XRD谱图,其中,谱线上的数值代表Mo、Si摩尔比。由图1可知,在不含Mo及Mo、Si摩尔比小于或等于8:100条件下合成的样品在2θ=0.99°、1.11°的位置均出现了明显的衍射峰,可分别归属于KIT-6分子筛的(211)(220)晶面的特征衍射峰,这表明骨架掺杂钼的Mo-KIT-6分子筛保持了原来的介孔结构并且有较好的有序度,而在Mo、Si摩尔比等于10:100的条件下合成的样品在小角度范围内没有明显的衍射峰,表明在引入大量钼的条件下没有形成有序的KIT-6分子筛介孔结构。随着Mo的引入,XRD衍射峰的峰位置向小角度偏移也证明钼掺杂进了KIT-6分子筛的骨架中。
图2为不同Mo、Si摩尔比条件下合成的Mo-KIT-6的紫外可见漫反射谱图。由图2可知,加入过渡金属钼后,在230nm及330nm处有两个明显的衍射峰,可分别 归属于四配位及六配位的钼。随着Mo掺杂量的增加,吸收峰强度也在增加,特别是330nm处的峰较230nm处的峰增加更为明显,说明聚合态的氧化钼物种在催化剂中比例逐渐增大,但350nm之后没有明显的吸收峰,这说明催化剂中没有晶相的氧化钼存在。
实施例2
本实施例提供了三种不同Mo、Si摩尔比条件下合成的丙烷选择氧化制丙烯醛和烯烃用催化剂(K/Mo-KIT-6催化剂),其制备方法包括以下步骤:
其中Mo-KIT-6分子筛的制备方法与实施例1完全相同;上述Mo、Si摩尔比分别为0.1:100、1:100、8:100;
称取0.0765g的硝酸钾,加入50g去蒸馏水,搅拌至完全溶解,得到硝酸钾溶液;
分别称取2g实施例1所制备的不同Mo掺杂量的Mo-KIT-6分子筛(选取Mo、Si的摩尔比为0.1:100,1:100,8:100的分子筛),分别各自加入2.2mg的上述硝酸钾溶液(因硝酸钾溶液的浓度较低,所以认为其密度近似于水的密度),在滴加过程中不断迅速搅拌;搅拌均匀后进行30min的超声分散处理,然后放入真空干燥箱中常温抽真空处理4h,所述真空干燥箱的真空度为0.01MPa,再放入烘箱中在100℃下干燥6h,最后放入马弗炉中以2℃/min的升温速率程序升温至550℃焙烧4h,得到K修饰不同钼含量的Mo-KIT-6分子筛,即丙烷选择氧化制丙烯醛和烯烃用催化剂(K/Mo-KIT-6催化剂),其中K与Si的摩尔比为0.1:100。
图3为实施例2中得到的丙烷选择氧化制丙烯醛和烯烃用催化剂(K/Mo-KIT-6催化剂)的UV-Vis谱图。
在图3中225nm和310nm处出现的吸收峰归属于高度分散的四配位钼物种和低聚的钼物种,而在大于320nm没有明显的吸收峰表明没有聚合的钼物种。相同钼载量的Mo-KIT-6催化剂加入K后,吸收峰相应的向低波长偏移,说明K的加入促进了钼氧化物的分散。一般活性金属的分散度高催化剂的活性会比较高,上述表征显示钼具有良好的分散状态,这说明催化剂有良好的活性。
实施例1得到的六种不同Mo、Si摩尔比的Mo-KIT-6催化剂和实施例2得到的丙烷选择氧化制丙烯醛和烯烃用催化剂K/Mo-KIT-6的相应物理化学参数如表1所示,表1中,a表示原料中的Mo/Si摩尔比,b表示Mo-KIT-6/K/Mo-KIT-6催化剂中Mo/Si的实际摩尔比,SBETc表示催化剂的BET比表面积,VTd表示催化剂的孔体积, Dpe表示催化剂的孔径。
从表1中可以看出,掺杂了钼的KIT-6催化剂与纯KIT-6相比,无论是其比表面积、孔容及孔径均变化不大,这说明材料的有序度较高,这与小角XRD数据一致。钼的实际最高掺杂量为3.8:100(Mo/Si摩尔比),说明在催化剂中活性组分含量较高。
表1
实施例3
本实施例提供了三种不同K、Si摩尔比条件下合成的丙烷选择氧化制丙烯醛和烯烃用催化剂(K/Mo-KIT-6催化剂),其与实施例2的区别之处仅在于硝酸钾溶液的浓度不同,其制备方法包括以下步骤:
分别称取0.0382g、0.1912g、0.3814g的硝酸钾,各自加入50g去蒸馏水,搅拌至完全溶解,得到三份不同浓度的硝酸钾溶液;
分别称取三等份2g实施例1所制备的Mo-KIT-6分子筛(选取Mo、Si的摩尔比为0.1:100的催化剂),分别各自加入2.2mg的上述硝酸钾溶液,在滴加过程中不断迅速搅拌;搅拌均匀后进行30min的超声振荡处理,然后放入真空干燥箱中常温抽真空处理4h,所述真空干燥箱的真空度为0.01MPa,再放入烘箱中在100℃下干燥6h,最后放入马弗炉中以2℃/min的升温速率程序升温至550℃焙烧4h,得到不同K载量修饰的Mo-KIT-6分子筛,即丙烷选择氧化制丙烯醛和烯烃用催化剂(K/Mo-KIT-6 催化剂),其中K与Si的摩尔比分别为0.05:100、0.25:100、0.5:100。
利用紫外可见漫反射光谱(UV-Vis)对实施例3提供的K/Mo-KIT-6催化剂进行检测,测定其表面活性金属离子的电荷转移以及d-d电子跃迁的性能,以表征金属离子的配位环境及价态信息,测试结果分别如图4所示。
图4为实施例3中得到的丙烷选择氧化制丙烯醛和烯烃用催化剂(K/Mo-KIT-6催化剂)的UV-Vis谱图。
由图4可以看出,添加碱金属钾后,催化剂在230nm附近的吸收峰明显增强,其归属于四配位的钼物种,而在330nm处的吸收峰明显减弱,表明碱金属的加入使六配位的钼向四配位的钼物种转移,使得四配位的钼含量高于六配位的钼。
图5为实施例3中得到的丙烷选择氧化制丙烯醛和烯烃用催化剂K/Mo-KIT-6的广角XRD图,从图5中可以看出,催化剂只有二氧化硅的衍射峰,没有钾氧化物等物质的衍射峰,说明K是以高分散形式存在的。
图6为实施例1中Mo、Si摩尔比为8:100的条件下合成的Mo-KIT-6的透射电镜图,图7为K、Mo、Si的摩尔比为0.1:8:100的K/Mo-KIT-6催化剂的透射电镜图。由图5及图6可知,Mo-KIT-6分子筛及K/Mo-KIT-6催化剂具有规则的有序孔道结构,孔道结构清晰可见,没有钾、钼的氧化物颗粒存在。透射电镜的表征结果证明合成的Mo-KIT-6分子筛及K/Mo-KIT-6催化剂保持了KIT-6介孔分子筛的三维孔道结构特征,并且钾、钼都是高分散的。
实施例4
本实施例提供了丙烷选择氧化制丙烯醛和烯烃用催化剂的丙烷选择氧化反应活性评价,即该催化剂在丙烷选择氧化制丙烯醛和烯烃反应中的应用。
对实施例1、2、3中制备的Mo-KIT-6分子筛及K/Mo-KIT-6催化剂的丙烷选择氧化反应活性进行评价测试,其中:
丙烷选择氧化反应的催化剂性能评价是在微型固定床反应装置上进行的,反应结束后使用气相色谱仪(SP-3420,北京分析仪器厂)对反应后的气体组成进行在线的定量分析。反应器为透明的固定床石英反应管,管内径为6mm,管壁厚为1mm。催化剂置于加热炉的恒温段位置,上下以石英棉固定。实验的温度控制采用精密的温度控制仪,加热炉采用程序控制升温。催化剂填装量为0.3g,原料气总流量为16mL·min-1(C3H8:O2:N2=1:1:6,体积比),反应气体的压力为0.4MPa,催化剂的样品粒度为60 目到100目。
1)反应器出口气相组成分析
以气相色谱仪(SP-3420,北京分析仪器厂)分离反应器出口的气相产物并检测。利用双FID氢火焰检测器分析反应器出口气体组成:用Porapack-Q柱(3m)分离丙烯、丙烷、乙烯、乙烷、乙醛、丙烯醛、丙酮和丙醇,并以氢火焰离子化检测器检测;以1m的TDX-01填充柱分离CO、CH4、CO2、HCHO、C2H4和C2H6,其中CO和CO2经过镍转化炉加氢转变为CH4后以氢火焰离子化检测器检测。
2)气相色谱分离检测条件
六通阀和进样器温度150℃,甲烷转化炉和检测器温度为380℃,色谱柱箱温度采用程序升温,初温70℃,保持15min,终温150℃,升温速率为40℃·min-1;色谱柱氮气载气稳流10mL·min-1。检测器的氢气流量30mL·min-1,空气流速300mL·min-1。
3)分析方法
采用面积归一法计算出丙烷转化率、产物选择性及收率;具体方法如下:
(1)产物的有效峰面积=色谱上获得的产物峰面积×反应物或产物的碳数×以丙烷为基准的相对校正因子,即Ai=A×Cn×fr;
(2)产物选择性=某一产物有效峰面积/总产物的有效峰面积之和
即Selectivity i%=Ai/∑Ai×100%;
(3)丙烷转化率=(丙烷原料有效峰面积-未反应的丙烷有效峰面积)/丙烷原料有效峰面积×100%;
(4)产物的收率=某一组分的选择性×丙烷转化率
即Yield i%=Selectivity i×Conversion C3H8×100%。
图8为实施例1提供的Mo-KIT-6分子筛(Mo、Si摩尔比为0.1:100、1:100、8:100)和实施例2提供的K/Mo-KIT-6(K、Mo、Si摩尔比为0.1:0.1:100、0.1:1:100、0.1:8:100)催化剂在催化丙烷选择氧化制丙烯醛和烯烃的反应中丙烷转化率结果图;
如图8所示,丙烷的转化率随着温度的升高不断增加,后保持不变或略有降低,在575℃左右丙烷转化率达到最高;对于不含K的分子筛催化剂,随着钼含量的增加,丙烷的转化率不断提高,在Mo/Si摩尔比为8:100时,转化率达到76.6%;加入钾后,丙烷的转化率均出现下降趋势,钼含量越高,下降越明显。对于钼含量较低的催化剂,其丙烷转化率均远远高于目前的文献报道,这也是最终产物丙烯醛的收率高 于其它文献报道的原因。
图9为实施例1提供的Mo-KIT-6分子筛(Mo、Si摩尔比为0.1:100、1:100、8:100)和实施例2提供的K/Mo-KIT-6(K、Mo、Si摩尔比为0.1:0.1:100、0.1:1:100、0.1:8:100)催化剂在催化丙烷选择氧化制丙烯醛和烯烃的反应中烯烃选择性的结果图;
如图9所示,烯烃的选择性随着温度的升高呈现出先增加后减小的趋势,Mo含量及K修饰对烯烃选择的影响与丙烷转化率表现出类似的作用。在不含K的催化剂上,钼含量较高时,在较低的温度下(500℃)表现出较高的烯烃的选择性(56.9%),K的引入同样引起烯烃选择性的下降,K、Mo、Si摩尔比为0.1:0.1:100的催化剂,在温度为575℃时,其烯烃的选择性达到最高也仅为48.4%。
图10为实施例1提供的Mo-KIT-6分子筛(Mo、Si摩尔比为0.1:100、1:100、8:100)和实施例2提供的K/Mo-KIT-6(K、Mo、Si摩尔比为0.1:0.1:100、0.1:1:100、0.1:8:100)催化剂在催化丙烷选择氧化制丙烯醛和烯烃的反应中丙烯醛选择性的结果图;
图11为实施例1提供的Mo-KIT-6分子筛(Mo、Si摩尔比为0.1:100、1:100、8:100)和实施例2提供的K/Mo-KIT-6(K、Mo、Si摩尔比为0.1:0.1:100、0.1:1:100、0.1:8:100)催化剂在催化丙烷选择氧化制丙烯醛和烯烃的反应中丙烯醛的收率结果图;
从图10及图11中可以看出,在不含K的催化剂中,高Mo含量的催化剂表现出了较好的丙烷选择氧化制丙烯醛的活性,丙烯醛收率达到19.4%,但加入K后其活性下降明显:但对于Mo含量较低的Mo-KIT-6催化剂,K的加入明显提高了丙烯醛的选择性,其中钼含量最少的K/Mo-KIT-6催化剂(K、Mo、Si的摩尔比为0.1∶0.1∶100)的丙烯醛选择性和收率效果最好,在625℃时丙烯醛选择性和摩尔收率分别达到31.5%和19.9%左右,这是目前报道的丙烷选择氧化制丙烯醛收率最好的结果之一。
图12为实施例3提供的K/Mo-KIT-6催化剂在催化丙烷选择氧化制丙烯醛和烯烃的反应中丙烷转化率结果图;
如图12所示,K的加入明显减小了丙烷的转化率,但在温度大约为575℃后,K、Si摩尔比为0.25:100的催化剂比不含K的催化剂表现出了更高的活性,在625℃时,丙烷的转化率达到74.1%。
图13为实施例3提供的K/Mo-KIT-6催化剂在催化丙烷选择氧化制丙烯醛和烯 烃的反应中烯烃选择性结果图;
如图13所示,在较低温度范围内,K的加入减小了烯烃的选择性,但温度大于575℃后,K改性的催化剂均表现出比无K催化剂更高的烯烃选择性,特别是K、Si摩尔比为0.1:100的催化剂,烯烃的选择性达到48.4%。
图14为实施例3提供的K/Mo-KIT-6催化剂在催化丙烷选择氧化制丙烯醛和烯烃的反应中丙烯醛选择性结果图;
图15为实施例3提供的K/Mo-KIT-6催化剂在催化丙烷选择氧化制丙烯醛和烯烃的反应中丙烯醛收率结果图;
如图14和15所示,在温度较低时,K的加入对丙烯醛选择性的影响不大,但温度大于525℃后,K的加入明显提高了丙烯醛的选择性,但K的负载量较高时(K=0.5),丙烯醛选择性提高较缓慢。综合考虑丙烯醛和烯烃的选择性,K、Mo、Si的摩尔比为0.25:0.1:100的K/Mo-KIT-6催化剂在丙烷选择氧化反应中有较高的活性及丙烯醛、烯烃的选择性,其中丙烯醛最高单程摩尔收率可达到26.0%,此时烯烃的摩尔收率也达到了27.5%。
实施例5
本实施例对实施例1中制备得到的Mo-KIT-6(Mo、Si摩尔比为8:100)催化剂的丙烷选择氧化反应活性进行评价测试,即该催化剂在丙烷选择氧化制丙烯醛和烯烃反应中的应用。该评价测试所用的装置和反应条件与实施例4的条件相同,只是在本实施例中催化剂的填装量分别为0.027g、0.053g、0.08g、0.16g、0.3g,其余条件不变,特别是反应气体总流量保持不变(原料气总流量为16mL·min-1)。因此,原料丙烷与催化剂的接触时间随着催化剂填装量的减小而缩短,实施例1提供的Mo-KIT-6催化剂在催化丙烷选择氧化制丙烯醛和烯烃的反应中,接触时间与反应活性的结果图如图16所示,从图16可以看出,随着催化剂填装量的减小,丙烷与催化剂的接触时间缩短,随着丙烷与催化剂接触时间的缩短,丙烯醛和丙烯的选择性都有较明显的提高。
以上所述的具体实施例,对本发明的目的、技术方案和有益效果进行了进一步详细说明,所应理解的是,以上所述仅为本发明的具体实施例而已,并不用于限定本发明的保护范围,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种铼聚合离子液体催化烯烃环氧化的方法与制造工艺
- 一种制备高纯度烯烃的催化剂的制备方法与制造工艺
- 稀土催化剂组合物及其制备方法和共轭二烯烃的聚合方法与制造工艺
- 一种催化剂的制备方法和制得的催化剂及其应用与制造工艺
- 一种用于烯烃聚合的催化剂体系及其应用的制造方法与工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分、催化剂及其应用的制造方法与工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺
- 用于烯烃聚合的催化剂组分及其制备方法和应用和用于烯烃聚合的催化剂及其应用与制造工艺