苯硼酸功能化的氧化石墨烯复合纳米材料及其制备和应用的制作方法
本发明涉及一种苯硼酸功能化的氧化石墨烯复合纳米材料及其制备和应用于糖蛋白的特异性富集。
背景技术:
硼亲和策略因其操作简单、无偏向性和不破坏糖型结构等特点,在糖基化蛋白质分离富集中得到越来越广泛的关注。但是传统的苯硼酸单体(PBA)亲和力低、与顺式邻二醇的结合能力弱导致对糖蛋白的选择性不高。据文献报道,一种新型的苯硼酸(2-羟甲基苯硼酸环状单酯及其衍生物)具有低pKa值(pKa=7.3),与顺式邻二醇的结合能力比传统苯硼酸更高,因此是一种非常有潜力的新型单体,已成功地应用于糖蛋白的富集(Chem.Commun.,2012,48,4115–4117;Small,2014,10,No.7,1379–1386)。硼酸功能化材料的制备多采用后修饰的方法,可用于键合硼酸基团的基质材料包括聚合物微球、琼脂糖凝胶、大孔硅胶以及磁性纳米颗粒等。纳米材料和聚合物比表面积有限,固载量有限;硅胶基质存在不可避免的非特异吸附。因此发展新型固定化基质材料十分必要。
氧化石墨烯(GO)作为石墨烯的一种重要衍生物,表面含有大量含氧基团,例如羟基、环氧和羧基。大量的含氧基团提高了氧化石墨烯化学稳定性和亲水性,也为制备氧化石墨烯复合材料提供反应位点。近年来,纳米粒子键合到氧化石墨烯表面形成的纳米复合物可以同时发挥二者的优异性质而成为一个热点研究,在催化、光电子材料、表面增强拉曼光散射、生物分析等领域具有很好的应用前景。其中,磁性氧化石墨烯由于结合了氧化石墨烯和磁性纳米颗粒的优点而成为一种新型功能材料,已经成功应用于染料和污染物吸附、药物固载和释放、蛋白质和DNA固载、适配体的固载等。因此,磁性氧化石墨烯纳米复合物可作为苯硼酸单体固定化的理想基质。
综上所述,我们将磁性氧化石墨烯纳米复合物作为固载基质来制备一种新型的苯硼酸功能化的氧化石墨烯复合纳米材料并用于糖蛋白的富集。
技术实现要素:
本发明的目的在于制备一种新型苯硼酸功能化的氧化石墨烯复合纳米材料,并用于糖蛋白的特异性富集。
为实现该目的,本发明采用的技术方案为:
苯硼酸功能化的氧化石墨烯复合纳米材料,是将四氧化三铁磁性纳米颗粒或外部包裹有高分子聚合物的四氧化三铁磁性纳米颗粒固载到氧化石墨烯上,随后对氧化石墨烯进行后修饰,最后引入苯硼酸功能单体。
所述的四氧化三铁磁性纳米颗粒粒径在100-300nm;或者在四氧化三铁磁性纳米颗粒外部包裹有高分子聚合物,其包裹壳层厚度在5-20nm,形成具有核壳结构的四氧化三铁磁性纳米颗粒;
将1-20质量份的四氧化三铁磁性纳米颗粒或外部包裹有高分子聚合物的四氧化三铁磁性纳米颗粒通过非共价作用物理吸附到1质量份的氧化石墨烯表面上;
1质量份氧化石墨烯上引入1-50质量份的高分子聚合物;
1质量份的高分子聚合物修饰的氧化石墨烯引入1-10质量份苯硼酸功能单体。
(1)合成粒径在100-300nm的四氧化三铁磁性纳米颗粒;或者在四氧化三铁磁性纳米颗粒外部包裹有高分子聚合物,其包裹壳层厚度在5-20nm,形成具有核壳结构的四氧化三铁磁性纳米颗粒;包裹的壳层为高分子聚合物即聚多巴胺或聚3,4-二羟基苯丙氨酸中的一种或二种以上。
(2)将1-20质量份的四氧化三铁磁性纳米颗粒或外部包裹有高分子聚合物的四氧化三铁磁性纳米颗粒通过非共价作用(π-π相互作用、氢键或离子键中的一种或二种以上)物理吸附到1质量份的氧化石墨烯表面上。
(3)将1质量份氧化石墨烯通过化学共价作用或者非共价作用(π-π相互作用、氢键或离子键中的一种或二种以上)引入1-50质量份树枝状或者超支化高分子聚合物;高分子聚合物包括聚乙烯亚胺、聚丙烯亚胺或聚酰胺胺中的一种或二种以上
(4)1质量份的高分子聚合物修饰的氧化石墨烯上通过共价键合引入1-10质量份苯硼酸功能单体。苯硼酸功能单体的共价键合具体包括下述中的一种或二种以上:通过氨基与羧基的酰胺化反应引入2-羧基苯硼酸(CPBA)、5-羧基-2-羟甲基苯硼酸(CBX)中的一种或二种以上;氨基与醛基的亲核加成反应引入4-甲酰基苯硼酸(FPBA);戊二醛作为偶联剂引入氨基苯硼酸(APBA)、5-氨基-2-羟甲基苯硼酸(ABX)中的一种或二种以上。
(5)将所述的苯硼酸功能化的氧化石墨烯复合纳米材料用于生物分析、生物技术领域内糖蛋白的特异识别、糖蛋白的富集。
本发明具有如下优点:
(1)制备简便、条件温和;其制备过程以水作为溶剂,环境友好。
(2)引入树枝状或者超支化的高分子聚合物,可以为后续苯硼酸功能单体键合提供更多的反应位点,提高材料的选择性。
(3)氧化石墨烯复合纳米材料作为固载基质具有比表面积高、非特异性吸附少等优点。
(4)非共价键合方法制备具有磁性的氧化石墨烯,不破坏氧化石墨烯性质。
附图说明
图1为Fe3O4/PDA透射电镜图。
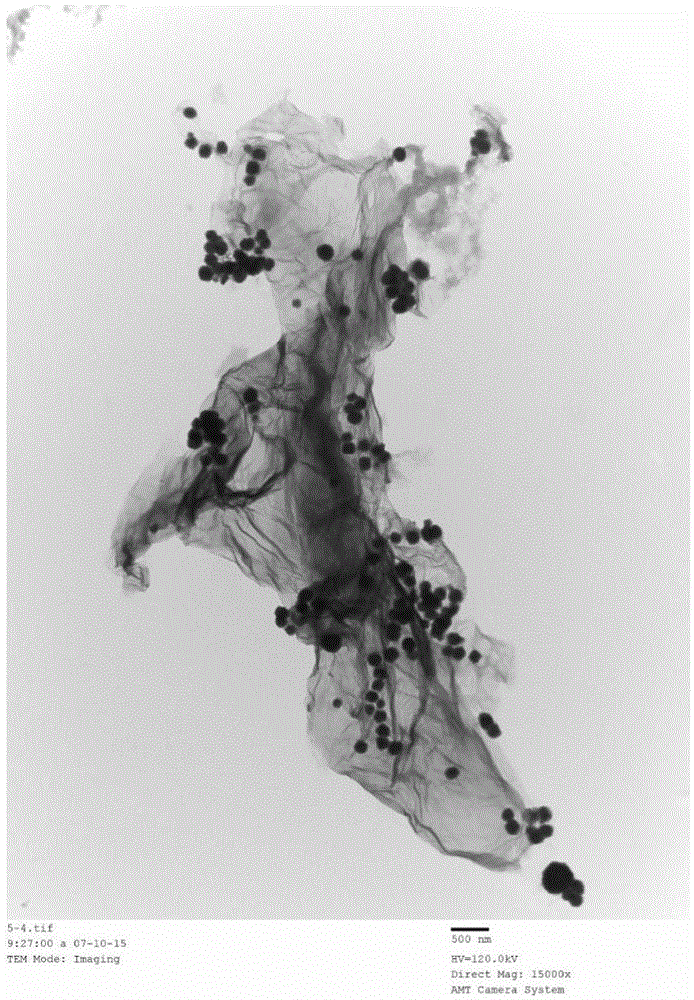
图2为Fe3O4/PDA/GO/BPEI透射电镜图。
图3为图2为Fe3O4/PDA/GO/BPEI/CBX透射电镜图。
图4为Fe3O4/PDA/GO/BPEI/CBX的X射线光电子能谱图。
图5为糖蛋白辣根过氧化物酶(HRP)与非糖蛋白牛血清白蛋白(BSA)经过Fe3O4/PDA/GO/BPEI/CBX富集后的MALDI-TOF图谱。
具体实施方式
实施例1
1.Fe3O4/PDA/GO/BPEI/CBX的制备:(1)称量1.08g的FeCl3·6H2O和0.2g的柠檬酸三钠,加入20ml的乙二醇,超声分散均匀。加入1.2g的无水乙酸钠,磁力搅拌至少30min。最后转移到反应釜,200℃反应10h。反应完毕,用去离子水和无水乙醇清洗产物各三次,真空干燥,得Fe3O4。(2)取100mg的Fe3O4,加入50ml 10mM pH=8.5的Tris-HCl缓冲盐溶液,超声分散均匀。快速加入100mg的多巴胺盐酸盐(DA),25℃水浴2h,得到Fe3O4/PDA。反应完毕,用去离子水和无水乙醇清洗产物各三次,真空干燥。(3)配制1mg/ml的GO水溶液,与分散均匀的Fe3O4/PDA水溶液混合,二者质量比为1:1,室温反应8h。反应完毕,用去离子水清洗产物6次,真空干燥。(4)配制1mg/ml的Fe3O4/PDA/GO水溶液,超声分散均匀。然后加入BPEI水溶液,其质量是Fe3O4/PDA/GO的10倍,室温反应10h。反应完毕,用去离子水清洗产物6次,真空干燥。(5)称量CBX单体15mg,加入0.1M pH=5.6MES(吗啉乙磺酸)15ml,超声使其溶解。加入64mg的EDC(1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐),19.8mg NHS(N-羟基琥珀酰亚胺)形成稳定的琥珀酰胺酯,40℃活化45min。加入30ml的0.1M Na2HPO4调节pH至中性,此时再加入15mg的Fe3O4/PDA/GO/BPEI,室温反应4h。反应完毕,用去离子水清洗产物6次,真空干燥,待用。
2.标准糖蛋白的富集:取100μg Fe3O4/PDA/GO/BPEI/CBX分散于200ul 50mM pH=9.0NH4HCO3中,加入HRP与BSA的混合物(质量比1:5),室温震荡2h。反应结束后,外加磁场分离弃去上清,三次缓冲盐淋洗非糖蛋白。然后用20ul洗脱液ACN/H2O/TFA(50:49:1)室温震荡1h洗脱HPR,洗脱馏分用于MALDI-TOF分析。
实施例2
1.Fe3O4/PDOPA/GO/PAMAM/CBX的制备:(1)称量1.08g的FeCl3·6H2O和0.2g的柠檬酸三钠,加入20ml的乙二醇,超声分散均匀。加入1.2g的无水乙酸钠,磁力搅拌至少30min。最后转移到反应釜,200℃反应10h。反应完毕,用去离子水和无水乙醇清洗产物各三次,真空干燥,得Fe3O4。(2)取100mg的Fe3O4,加入50ml 10mM pH=8.5的Tris-HCl缓冲盐溶液,超声分散均匀。快速加入100mg的3,4-二羟基苯丙氨酸(DOPA),25℃水浴2h,得到Fe3O4/PDOPA。反应完毕,用去离子水和无水乙醇清洗产物各三次,真空干燥。(3)配制1mg/ml的GO水溶液,与分散均匀的Fe3O4/PDOPA水溶液混合,二者质量比为1:1,室温反应8h。反应完毕,用去离子水清洗产物6次,真空干燥。(4)配制1mg/ml的Fe3O4/PDOPA/GO水溶液,超声分散均匀。然后加入PAMAM溶液,其质量是Fe3O4/PDOPA/GO的10倍,室温反应10h。反应完毕,用去离子水清洗产物6次,真空干燥。(5)称量CBX单体15mg,加入0.1M pH=5.6MES 15ml,超声使其溶解。加入64mg的EDC,19.8mg NHS形成稳定的琥珀酰胺酯,40℃活化45min。加入30ml的0.1M Na2HPO4调节pH至中性,此时再加入15mg的Fe3O4/PDOPA/GO/PAMAM,室温反应4h。反应完毕,用去离子水清洗产物6次,真空干燥,待用。
2.标准糖蛋白的富集:取100μg Fe3O4/PDOPA/GO/PAMAM/CBX分散于200ul 50mM pH=9.0NH4HCO3中,加入HRP与BSA的混合物(质量比1:5),室温震荡2h。反应结束后,外加磁场分离弃去上清,三次缓冲盐淋洗非糖蛋白。然后用20ul洗脱液ACN/H2O/TFA(50:49:1)室温震荡1h洗脱HPR,洗脱馏分用于MALDI-TOF分析。
实施例扩充。
实施例3
1.Fe3O4/PDA/GO/BPEI/CPBA的制备:(1)称量1.08g的FeCl3·6H2O和0.2g的柠檬酸三钠,加入20ml的乙二醇,超声分散均匀。加入1.2g的无 水乙酸钠,磁力搅拌至少30min。最后转移到反应釜,200℃反应10h。反应完毕,用去离子水和无水乙醇清洗产物各三次,真空干燥,得Fe3O4。(2)取100mg的Fe3O4,加入50ml 10mM pH=8.5的Tris-HCl缓冲盐溶液,超声分散均匀。快速加入100mg的多巴胺盐酸盐,25℃水浴2h,得到Fe3O4/PDA。反应完毕,用去离子水和无水乙醇清洗产物各三次,真空干燥。(3)配制1mg/ml的GO水溶液,与分散均匀的Fe3O4/PDA水溶液混合,二者质量比为1:1,室温反应8h。反应完毕,用去离子水清洗产物6次,真空干燥。(4)配制1mg/ml的Fe3O4/PDA/GO水溶液,超声分散均匀。然后加入BPEI水溶液,其质量是Fe3O4/PDA/GO的10倍,室温反应10h。反应完毕,用去离子水清洗产物6次,真空干燥。(5)称量CPBA单体15mg,加入0.1M pH=5.6MES 15ml,超声使其溶解。加入64mg的EDC,19.8mg NHS形成稳定的琥珀酰胺酯,40℃活化45min。加入30ml的0.1M Na2HPO4调节pH至中性,此时再加入15mg的Fe3O4/PDA/GO/BPEI,室温反应4h。反应完毕,用去离子水清洗产物6次,真空干燥,待用。
2.标准糖蛋白的富集:取100μg Fe3O4/PDA/GO/BPEI/CPBA分散于200ul50mM pH=9.0NH4HCO3中,加入HRP与BSA的混合物(质量比1:5),室温震荡2h。反应结束后,外加磁场分离弃去上清,三次缓冲盐淋洗非糖蛋白。然后用20ul洗脱液ACN/H2O/TFA(50:49:1)室温震荡1h洗脱HPR,洗脱馏分用于MALDI-TOF分析。
实施例4
1.Fe3O4/PDA/GO/PAMAM/FPBA的制备:(1)称量1.08g的FeCl3·6H2O和0.2g的柠檬酸三钠,加入20ml的乙二醇,超声分散均匀。加入1.2g的无水乙酸钠,磁力搅拌至少30min。最后转移到反应釜,200℃反应10h。反应完毕,用去离子水和无水乙醇清洗产物各三次,真空干燥,得Fe3O4。(2)取100mg的Fe3O4,加入50ml 10mM pH=8.5的Tris-HCl缓冲盐溶液,超声分散均匀。快速加入100mg的多巴胺盐酸盐,25℃水浴2h,得到Fe3O4/PDA。反应完毕,用去离子水和无水乙醇清洗产物各三次,真空干燥。(3)配制1mg/ml的GO水溶液,与分散均匀的Fe3O4/PDA水溶液混合,二者质量比为1:1,室温反应8h。反应完毕,用去离子水清洗产物6次,真空干燥。(4)配制1mg/ml的Fe3O4/PDA/GO水溶液,超声分散均匀。然后加入PAMAM水溶液,其质量是Fe3O4/PDA/GO的10倍,室温反应10h。反应完毕,用去离子水清洗产物6次,真空干燥。(5)称量200mg Fe3O4/PDA/GO/PAMAM分散于40ml的无水甲醇中,加入400mg FPBA 单体,机械搅拌24h。反应过程中每隔四个小时加入100mg的氰基硼氢化钠。反应完毕,用去离子水清洗产物6次,真空干燥,待用。
2.标准糖蛋白的富集:取100μg Fe3O4/PDA/GO/PAMAM/FPBA分散于200ul 50mM pH=9.0NH4HCO3中,加入HRP与BSA的混合物(质量比1:5),室温震荡2h。反应结束后,外加磁场分离弃去上清,三次缓冲盐淋洗非糖蛋白。然后用20ul洗脱液ACN/H2O/TFA(50:49:1)室温震荡1h洗脱HPR,洗脱馏分用于MALDI-TOF分析。
- 还没有人留言评论。精彩留言会获得点赞!