一种催化剂及由合成气一步法直接制备低碳烯烃的方法与流程
本发明属于制备低碳烯烃领域,具体涉及一种由合成气一步法制备低碳烯烃的方法及其催化剂。
背景技术:
低碳烯烃包括乙烯、丙烯、丁烯,是合成塑料、纤维和各类化工材料的重要原料,随着国民经济的增长,其消费需求也越来越快。目前,低碳烯烃主要由石脑油裂解的石油化工路线生产获得。随着全球石油资源日渐消耗和居高不下的原油价格,尤其对石油资源匮乏的我国而言,每年超过近60%的石油消耗量依赖进口,我国低碳烯烃生产的原料70%为原油炼制得到的石脑油和轻烃,寻求一种可以替代的工艺路线,开发利用由煤、生物质等非油基碳资源制备低碳烯烃的方法,具有重要的社会意义和战略意义。
我国煤炭资源丰富,以煤炭为原料,经过气化得到合成气(即CO和H2的混合气),再将合成气转化制甲醇,甲醇制烯烃(MTO)的工艺成熟,已经步入工业化。催化剂为改性的分子筛SAPO-34或ZSM-5,甲醇转化率接近100%,乙烯和丙烯选择性85-90%,仅有少量的C5+以上长碳链碳氢化合物生成。2010年8月,世界首套基于大连化物所具有自主知识产权的甲醇制烯烃(DMTO)技术的工业装置成功实现商业运转,标志着我国煤制烯烃新兴产业取得了里程碑式的进展。2015年4月,同样采用大连化物所核心技术的浙江兴兴新能源60万吨/年MTO装置投入商业运营,是我国第八套大型煤基甲醇制烯烃工业装置。
与经甲醇制烯烃的间接途径相比,从合成气一步法直接制备烯烃的路线则具有工艺简单,设备投资少的特点,国内外科研院所以及企业投入了大量了资源,开展合成气一步法制低碳烯烃工艺路线的研究。中国石化上海石油化工研究院对合成气由费托路线直接制取低碳烯烃技术的经济性进行了评估[杨学萍,董丽,化工进展31(2012)1726-1731]。当油价为110美元/桶,而煤价不高于520元/吨,烯烃在总产品中组成为30%时,煤基合成气直接制低碳烯烃的销售成本就比较有优势。若能进一步提高低碳烯烃在总产品中在含量提高至40%以上,其经济性优势将更加明显。
由合成气直接转化制低碳烯烃,可以通过传统费托反应路径获得。该反应路径中,通常认为是由CO在催化剂表面解离吸附,经历加氢生成表面吸附的CHx中间物种,然后单个碳物种逐个经表面催化插入经历聚合反应实现碳链增长,生成不同碳数的碳氢化合物。经典的产物分布遵循Anderson-Schultz-Flory(ASF)分布方程,其数学表达式为:ln(Wn/n)=nlnP+ln[(1-P)2/P],式中,Wn为碳数n的质量分数,P为链增长几率,(1-P)为碳链终结几率,聚合度D=1/(1-P),一旦P确定,整个产物分布随之确定,不同碳数的碳氢化合物分布宽,可预测的C2-C4烃类选 择性最高不超过58%,汽油馏分C5-C11最高选择性为45%,同时生成大量甲烷和高碳烷烃。为此,如何选择性地生成低碳烯烃一直是该领域难以克服的挑战。国内外的研究人员做了很多努力,通过修饰催化剂结构和组成,来调变其中关键步骤的相对反应速率,如甲烷化、加氢、低碳烯烃的二次反应、碳链增长等。
其中Fe基催化剂具有价廉易得、活性高和低碳烯烃选择性高等优点,是最具潜力的合成低碳烯烃催化剂。研究人员通过添加不同组成的添加剂,如碱金属K、Na和过渡金属Mn、Cu等,可以有效改善低碳烯烃的选择性。德国鲁尔化学公司开发了多组分助剂促进的Fe-Zn-Mn-K催化剂,用于低碳烯烃的合成[12]。北京化工大学张敬畅等报道以草酸铁为前驱体制备的Fe-Mn-K/AC催化剂在空速600h-1,15bar和320℃下,CO的转化率高达97%,C2=-C4=在碳氢化合物中的选择性为68%[张敬畅,卫国宾,曹维良,催化学报24(2003)259-264],超过了由ASF分布模型预测的C2-C4烃的选择性。催化剂的载体通过与Fe物种的相互作用,也对产物的选择性起重要的修饰作用。荷兰de Jong课题组最近报道以碳纳米纤维(CNF)和α-Al2O3为载体,以柠檬酸铁胺为前驱体制备的12wt%Fe催化剂,在低压(1bar),350℃,H2/CO=1条件下,反应15小时,CO转化率为0.5-1.0%,低碳烯烃在碳氢化合物中的选择性为60%[H.M.T.Galvis,J.H.Bitter,C.B.Hhare,M.Ruitenbeek,A.L.Dugulan,K.P.de Jong,Science 335(2012)835-838]。同样的催化剂在H2/CO=1,20bar,空速1500h-1下,获得了70-88%的转化率,FTY分别为2.98×10-5mol CO/gFe·s和1.35×10-5mol CO/gFe·s,CO2的选择性为42-46%,其中低碳烯烃在碳氢化合物中的选择性为52-53%。随后他们发现,催化剂中的微量0.03%S,0.2%左右的Na,对反应活性和低碳烯烃的选择性具有明显的促进作用[H.M.T.Galvis,A.C.J.Koeken,J.H.Bitter,T.Davidian,M.Ruitenbeek,A.I.Dugulan,K.P.de Jong,J.Catal.303(2013)22-30]。中国科学院大连化物所对活性炭载体作了系统深入的研究,发现活性炭负载的铁催化剂上产物偏离ASF分布[沈俭一,林励吾,章素,梁东白,燃料化学学报19(1991)289-297;马文平,丁云杰,罗洪原等,催化学报22(2001)279-282]。此外,催化剂的制备方法和工艺,如焙烧过程、还原条件等,可以直接影响活性组分的分散和尺寸,从而调变反应的活性和产物的选择性。北京化工大学采用超临界流体组合技术(即化学沉淀、类凝胶、超临界干燥的方法)使活性组分Fe和助剂高度分散,制备了纳米级Fe基催化剂,CO的转化率大于96%,低碳烯烃在碳氢化合物中的选择性大于54%[北京化工大学,一种用于合成气制低碳烯烃的纳米催化剂及制备方法:中国,101396662[P].2009-04-01]。
有研究人员设法将费托反应与其它工艺进行组合,如裂解反应[J.L.Park,Y.J.Lee,K.W.Jun,J.W.Bae,N.Viswanadham,Y.H.Kim,J.Ind.Eng.Chem.15(2009)847-853],通过双床层反应器,首先在第一个反应器Fe-Cu-Al催化剂上300℃、10atm、GHSV=3600h-1下进行费托反应,再经过第二个反应器500℃下ZSM-5裂解催化剂床层,将大量的C5+产物裂解为低碳烯烃,由此得到的碳氢化合物产物中低碳烃的选择性为52%,低碳烯烃在总产品中的选择性为28%。
此外,由于合成气直接制甲醇和甲醇制烯烃的过程相对成熟,人们对这两个过程的耦合也做了大量的尝试。Xu等将CuO-ZnO-Al2O3与ZSM-5混合,得到的催化剂,在合成气转化反应中得到的产物主要为二甲醚[M.Xu,J.H.Lunsford,D.W.Goodman,A.Bhattacharyya,Appl.Catal.A.General 149(1997)289;D.Mao,W.Yang,J.Xia,B.Zhang,Q.Song,Q.Chen,J.Catal.230(2005)140]。Erena等将CuO/ZnO/Al2O3等多组分金属复合物与ZSM-5分子筛混合,催化合成气转化,得到的产物主要为汽油[J.Erena,J.M.Arandes,J.Bilbao,A.G.Gayubo,H.I.De Lasa,Chemical Engineering Science 2000,55,1845;J.Erena,J.M.Arandes,R.Garona,A.G.Gayubo,J.Bilbao,Journal of Chemical Technology and Biotechnology2003,78,161]。
本发明提供了一种合成气直接转化制低碳烯烃的方法及其催化剂,得到的碳氢化合物中低碳烃,即含有2个到4个碳原子的烃类产物的选择性高达60~95%,其中低碳烯烃包括乙烯、丙烯和丁烯的选择性占50~85%。
技术实现要素:
针对上述问题,本发明提供了一种由合成气一步法直接转化制低碳烯烃的方法及其催化剂。该方法通过一种复合催化剂材料,原料分子CO在具有缺陷位的金属复合物表面活化,吸附的另外的CO与解离生成的氧原子反应生成CO2,由于另一个原料分子氢不能与解离的氧原子生成水,避免了大量高值的氢的消耗,这样该反应原料的氢碳比就可以非常小,理论上可以为H/C=0.5,在这种情况下,该工艺可以直接利用煤气化炉出口的混合气,可以减除高能耗的水煤气变换工段;另一方面,在复合物表面生成的CHx中间体不能在复合物表面停留,会以自由基的形式脱附到气相中,并进入多孔材料的孔道中,进行限域的链增长,选择生成目标烃类分子,实现了一步法直接催化转化合成气,高选择性地生成低碳烯烃,这样过程不仅突破了传统费托反应路径中产物ASF分布的限制,高选择性地获得低碳烯烃,而且摒弃了额外的甲醇合成过程,以及水煤气变换过程。低碳烯烃在碳氢化合物中的选择性高达50~85%,一氧化碳的单程转化率为10~50%。该方法具有工艺简单,设备投资少的特点,可以预见,该方法可以显著降低低碳烯烃的生产成本,具有重要的应用价值。
本发明的技术方案为:
所述催化剂为多组分金属复合物和多级孔结构的无机固体酸构成的复合材料;无机固体酸具有微孔、介孔和大孔的多级孔结构;金属复合物分散在无机固体酸表面或孔道中;
按催化剂的总质量为100%计,所述多组分金属复合物在整个催化剂中的含量为10wt.%~75wt.%,优选25wt.%-75wt.%;
所述多级孔结构的无机固体酸是由无机固体酸二次粒子组成,无机固体酸二次粒子大小为100nm-500μm,优选为150nm-100μm;
所述无机固体酸二次粒子是由大小为5-200nm(优选20-120nm)的无机固体酸晶粒堆积形成的;
所述无机固体酸二次粒子具有三维三级孔道结构,包括一级孔,二级孔,三级孔三种孔道;
所述一级孔是孔道直径小于2nm范围的微孔孔道,微孔位于无机固体酸晶粒内部;
所述二级孔是孔道直径2nm-50nm的介孔孔道,优选为2-15nm,二级孔是由无机固体酸晶粒堆积而成,位于无机固体酸二次粒子内部及三级孔的孔壁上;
所述三级孔是孔道直径分布大于50nm的大孔孔道,是无机固体酸二次粒子堆积而成;
所述三种孔道之间相互连接贯通,构成三维三级孔道结构,二级孔可位于相邻三级孔孔壁,一级孔可位于相邻的二级孔和/或三级孔孔壁;
所述的多级孔结构的无机固体酸由N2物理吸附测定的BET比表面积为100~1200m2/g,孔容为0.25~0.80ml/g;按照比表面积计算,微孔占10-65%,介 孔占20-75%,大孔占15-70%;优选的微孔的比表面积占10-60%,介孔比表面积占20-70%,大孔占20-70%;更优选为微孔的比表面积占10-50%,介孔比表面积占总比表面积的30-70%,大孔占20-60%。
所述的多组分金属复合物中的金属为两种或三种以上的金属元素,优选为两种至五种金属元素;所述金属复合物包括金属氧化物以及金属、金属碳化物、金属氮化物、和金属无机酸盐中的一种或二种以上;
所述金属元素包括必要金属元素以及其它元素,其中必要金属元素为Zn或Co或Cr或Mn;
若所述必要金属元素为Zn,则其它元素为Li、Na、Mg、Al、K、Ca、Sr、Ba、Ti、V、Cr、Mn、Fe、Co、Cu、Ga、Ge、Zr、Mo、Pd、Ag、Cd、In、Sn、Cs、La、Ce中的一种或二种以上;其它元素优选为Al、K、Ti、V、Cr、Mn、Co、Cu、Ce、Pd中的一种或二种以上,其它元素更优选为Al、Ti、Cr、Mn、Co、Cu、Pd、Ce中的一种或二种以上;
若所述必要金属元素为Co时,其它元素为Li、Na、Mg、Al、K、Ca、Sr、Ba、Ti、V、Cr、Mn、Fe、Cu、Zn、Ga、Ge、Zr、Mo、Pd、Ag、Cd、In、Sn、Cs、La、Ce中的一种或二种以上;其它元素优选为Al、K、Ti、V、Cr、Mn、Cu、Zn、Ce、Pd中的一种或二种以上,其它元素更优选为Al、Ti、Cr、Mn、Cu、Zn、Pd、Ce中的一种或二种以上;
若所述必要金属元素为Cr,则其它元素为Li、Na、Mg、Al、K、Ca、Sr、Ba、Ti、V、Mn、Fe、Co、Cu、Zn、Ga、Ge、Zr、Mo、Pd、Ag、Cd、In、Sn、Cs、La、Ce中的一种或二种以上;其它元素优选为Al、K、Ti、V、Mn、Co、Cu、Zn、Ce、Pd中的一种或二种以上,其它元素更优选为Al、Ti、Mn、Co、Cu、Zn、Pd、Ce中的一种或二种以上;
若所述必要金属元素为Mn,则其它元素为Li、Na、Mg、Al、K、Ca、Sr、Ba、Ti、Cr、Fe、Co、Cu、Zn、Ga、Ge、Zr、Mo、Pd、Ag、Cd、In、Sn、Cs、La、Ce中的一种或二种以上;其它元素优选为Al、K、Ti、V、Cr、Co、Cu、Zn、In、La、Mo、Ce、Pd中的一种或二种以上,其它元素更优选为Al、Ti、Cr、Co、Cu、Zn、Pd、Ce中的一种或二种以上;
所述的多组分金属复合物中,金属氧化物的含量为5~90%,金属和金属碳化物和金属氮化物中的一种或两种以上的总质量含量可以是小于或者等于10%,,金属无机酸盐的含量为10~95%;优选金属氧化物的含量为30-90%,金属和金属碳化物和金属氮化物中的一种或两种以上的总质量含量小于5%,金属无机酸盐的含量为10-70%;
所述的多组分金属复合物中必要元素Zn或Co或Cr或Mn与其他元素的总摩尔比为(0.1~5.0):1,优选摩尔比为0.2~3.5;
所述无机酸盐是由阳离子与阴离子组成,其中Li、Na、Mg、Al、K、Ca、Sr、Ba、Ti、V、Cr、Mn、Fe、Co、Cu、Zn、Ga、Ge、Zr、Mo、Pd、Ag、Cd、Sn、Cs、Ce中的一种或二种以上可以以阳离子形式存在,而Al、Si、V、Cr、Mn、Fe、Co、Zn、Mo、Ti、Zr、Ce、Ga、In、Ge也可以以ZnO22-、Al2O42-、SiO32-、SiO44-、TiO32-、TiO33-、VO3-、VO32-、CrO42-、Cr2O42-、Mn2O42-、Fe2O42-、Co2O42-、Ni2O42-、Fe(CN)63-、Fe(CN)64-、MoO42-、TiO32-、ZrO32-、CeO32-、Ga2O42-、In2O42-、GeO32-、GeO44-、SrO34-中的一种或二种以上阴离子形式存在,阳离子与阴离子组成无机酸盐,无机酸盐组成如表1所列;且构成阳离子和构成阴离子的金属元素不同。
所述金属复合物是由权利要求2中所述金属氧化物以及金属、金属碳化物、金属氮化物、金属无机酸盐中一种或两种以上的晶粒相互均匀分散形成,其晶粒大小为0.5-20nm,优选1-10nm;同时这些晶粒进一步堆积形成二次粒子,二次粒子大小为10nm-200μm,二次粒子中晶粒堆积形成的孔道直径为2-20nm,优选为5-15nm。
所述的无机固体酸由H、O为必要元素,以及Al、Si、P、Zr、Ti、Ge中的一种或两种以上元素组成,形成由硅氧氢元素组成的无机固体酸、由硅铝氧氢元素组成的无机固体酸、由硅铝磷氧氢元素组成的无机固体酸、由钛硅氧氢元素组成的无机固体酸、由锆硅氧氢元素组成的无机固体酸、由锗硅氧氢元素组成的无机固体酸、由锗铝磷氧氢元素组成的无机固体酸中的一种或二种以上。
所述的多级孔结构的无机固体酸具有酸性特征;
所述的酸性位分布于权利要求1所述的三维三级孔道;
按照NH3-TPD定义酸性强弱,所述固体无机酸包含弱酸、中强酸、强酸三种酸性;
该NH3-TPD是根据NH3的脱附峰位置,所述脱附峰的位置是指在标准测试条件下,在样品质量w与载气流速f比值(w/f)=100g·h/L,10℃/min升温速度的测试条件下,TCD记录脱附NH3的热导信号,绘制脱附曲线,根据曲线峰位置顶点将所述无机固体分为三种酸性强度;
所述弱酸是NH3脱附温度小于275℃的酸性位;
所述中强酸是NH3脱附温度在275-500℃的酸性位;
所述强酸是NH3脱附温度大于500℃的酸性位;
所述无机固体酸中,中强酸位点的量是0.06-10mol/kg;优选1-10mol/kg;
所述的多组分金属复合物和多级孔结构的无机固体酸构成复合材料;多组分金属复合物中尺寸在0.5-10nm的粒子可以位于多级孔结构的无机固体酸的微孔、介孔或者大孔孔道内以及无机固体酸外表面,多组分金属复合物中尺寸在10-200nm的二次粒子可以位于多级孔结构的无机固体酸的大孔和介孔的孔道内或者无机固体酸外表面,尺寸大于200nm的金属复合物二次粒子分散在多级孔结构的无机固体酸外表面,多组分金属复合物的外表面与无机固体酸的大孔和介孔的内表面及无机固体酸二次粒子的外表面堆积形成活性孔;所述活性孔连接金属复合物介孔与多级孔结构的无机固体酸中的三维三级孔道,使得所有孔道连通复合。
所述的多级孔结构的无机固体酸制备方法包括软模板水热法、硬模板水热法或结晶控制水热法;在软模板水热法、硬模板水热法或结晶控制水热法的老化操作中,使用恒温搅拌老化处理有利于形成多级孔结构的无机固体酸;
所述结晶控制水热法,快速老化,并在晶化过程中控制搅拌速率和温度,避免分子筛晶粒过度长大,促进小晶粒的形成,使更多的小晶粒之间产生晶间介孔,获得多级孔结构的无机固体酸,同时通过调变老化及晶化条件可以同时调变权利要求5所述的酸性强弱及中强酸的量;具体步骤包括:均匀溶胶分散液的制备、老化、晶化、洗涤、干燥和焙烧;所述的均匀溶胶分散液的制备是将无机固体酸所含有元素的前驱体按所需比例,称量后投入含有水的容器中,在室温下进行搅拌分散,同时加入有机微孔模板剂制得液相分散体系;所述老化方法是将配制好的液相分散体系在温度控制在20-60℃,时间10min-24h,搅拌速率50-1000rpm;晶化过程中搅拌速率50-500rpm,温度120-250℃,时间12h-10day;洗涤方法可以是过滤洗涤或离心洗涤,过滤洗涤应保证洗涤结束时滤液pH是6.5-7.5,离心 洗涤应保证洗涤结束时上清液是6.5-7.5;所述干燥过程,温度在100-150℃,时间大于12h;所述焙烧过程,温度在500-650℃,时间1-8h;
所述软模板水热法,是利用有机介孔模板剂,使用水热法合成多级孔结构的无机固体酸,具体步骤涉及均匀溶胶分散液的制备,老化,晶化,洗涤,干燥和焙烧;所述的均匀溶胶分散液的制备是将无机固体酸所含有元素的前驱体按所需质量比,称量后投入含有水的容器中,在室温下进行搅拌分散,同时加入有机微孔模板剂及有机介孔模板剂(其质量比为0.3~0.8),制得液相分散体系,老化过程中温度控制在20-60℃,时间10min-24h,搅拌速率50-1000rpm;晶化过程中搅拌速率50-500rpm,温度120-250℃,时间12h-10day;洗涤方法可以是过滤洗涤或离心洗涤,过滤洗涤应保证洗涤结束时滤液pH是6.5-7.5,离心洗涤应保证洗涤结束时上清液是6.5-7.5;所述干燥过程,温度在100-150℃,时间大于12h;所述焙烧过程,温度在500-650℃,时间1-8h;
所述硬模版水热法,是可以在650℃下被氧化的纳米碳材料、SBA系列、M41S系列、HMS系列、MSU系列、KIT系列、FDU系列、AMS系列、HOM系列、MCM-41介孔分子筛中的一种或两种以上作为硬模板,使用水热法合成多级孔结构的无机固体酸,具体步骤涉及均匀溶胶分散液的制备,老化,晶化,洗涤,干燥和焙烧;所述的均匀溶胶分散液的制备是将无机固体酸所含有元素的前驱体按所需质量比,称量后投入含有水的容器中进行搅拌分散,同时加入有机微孔模板剂及硬模板材料(其质量比例为0.3~0.8)制得液相分散体系,抑制晶粒长大,促进小晶粒形成介孔;温度控制在20-60℃,时间10min-24h,搅拌速率50-1000rpm;晶化过程中搅拌速率50-500rpm,温度120-250℃,时间12h-10day,洗涤方法可以是过滤洗涤或离心洗涤,过滤洗涤应保证洗涤结束时滤液pH是6.5-7.5,离心洗涤应保证洗涤结束时上清液是6.5-7.5;所述干燥过程,温度在100-150℃,时间大于12h;所述焙烧过程,温度在500-700℃,时间1-8h;
所述均匀溶胶分散液中前驱体是水玻璃、硅溶胶、超微SiO2、白炭黑、硅酸钠、TMOS、硅酸酯、四氯化硅、高岭土、硝酸铝、硫酸铝、铝酸钠、薄水铝石、拟薄水铝石、三水铝石、三水异丙醇铝、三氯化铝、氢氧化铝、烷氧基铝、超微粉末铝、氧化铝、磷酸、磷酸铝、磷酸钠、氧化锆、硝酸锆、磷酸锆、硅酸锆、四氯化钛、钛酸丁酯、氧化钛、氧化锗、硝酸锗、氯化锗、三氯甲基锗、四乙基锗中的一种或两种以上;
所述有机微孔模板剂是TMA、TPA、TEA、TEAOH、DDO、TBA、TQA、乙二胺、吡咯烷、胆碱、PDN、RDN、PDA、季戊四醇、羟酸、丙基硫醛、三丙胺、DABCO、二正丙胺、叔丁胺、异丙胺、奎宁环、新戊胺、三乙醇胺、二环己胺、N,N-二甲基苄胺、N,N-二甲基醇胺、N,N-二乙基乙醇胺、2-甲基吡啶、哌啶、吗啡中的一种或两种以上;
所述有机介孔模板剂是CTBA、CTBABr、十二烷基磷酸酯、长链伯胺、聚氧乙烯、聚氧丙烯、聚氧乙烯嵌段醚共聚物、P123、明胶与戊二醛交联物、TPHAC中的一种或两种以上;
所述溶胶前驱体和有机模板剂(包括有机微孔模板剂和有机介孔模板剂)以及水的质量比为,溶胶前驱体:有机模板剂:水=(20-65):(15-50):100。
所述催化剂中多组分金属复合物和多级孔结构的无机固体酸的复合方式包括包覆生长法,超声辅助的化学复合法,或物理复合法;
所述的包覆生长法,是在多组分金属复合物上,通过权利要求6所述的方法,在多组分金属复合物表面生长出一层覆盖率至少80%以上或者完全覆盖的多级孔结构的无机固体酸,即先制备均匀溶胶分散液,再将多组分金属复合物分散于均匀溶胶分散液中,再经过老化、晶化、洗涤,干燥和焙烧得到包覆有多级孔结构的无机固体酸的多组分金属复合物;多组分金属复合物中,由Mg、Al、Ca、Sr、Ba、Ti、V、Cr、Mn、Fe、Co、Cu、Zn、Zr、Mo、Pd、Ag、Cd、In、Sn、La、Ce元素组成的金属氧化物和无机酸盐由共沉淀法、浸渍法、或超声离子交 换法制备;由Li、Na、K、Ga、Ge、Cs元素组成的金属氧化物和无机酸盐由浸渍法、或超声离子交换法制备;Al、Fe、Co、Cu、Zn、Mo、Pd元素还可采用化学气相沉积法或者浸渍法制备;
所述的超声辅助的化学复合法,是在多级孔结构的无机固体酸上通过超声共沉淀法,化学气相沉积法或超声离子交换法引入多组分金属复合物复合得到;
所述的物理复合法,是通过球磨,机械混匀法,摇床,振荡混合的机械化学法对多级孔结构的无机固体酸和多组分金属复合物进行复合。
所述的超声共沉淀法,指将多组分金属复合物所需两种或三种以上的金属前驱体溶解于溶剂中,得到两种或三种以上的金属阳离子溶液,然后将多级孔结构的无机固体酸在超声搅拌条件下加入到上述溶液中,超声方法可以是bath超声或Tip超声,超声功率为1-20W/(ml样品),频率40KHz-80MHz,沉淀温度2℃-80℃,时间2min-2h,搅拌速率50-1000rpm,混合均匀之后,按所需质量比加入沉淀剂,得到均匀的沉淀物,再经干燥、焙烧,干燥温度是80-150℃,时间大于12h,焙烧温度是300-650℃,时间1-3h;
所述超声共沉淀法中,金属前驱体选自硝酸盐、甲酸盐、醋酸盐、卤化物、羰基化合物、C1~C5有机酸盐中的一种或两种以上;沉淀剂选自碳酸铵、碳酸氢铵、碳酸锂、碳酸氢锂、碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾、碳酸镁或草酸中的一种或两种以上,优选为碳酸铵、碳酸氢铵、碳酸氢钠、碳酸氢钾,最佳为碳酸铵、碳酸氢铵、碳酸氢钠;沉淀温度控制在2~80℃的范围内,干燥温度为80~150℃,时间大于12h,焙烧温度为300~650℃,时间1-3h,焙烧气氛可以是流动的空气或者静置空气;
所述超声离子交换法是指将多组分金属复合物所需两种或三种以上的金属前驱体溶解于溶剂中,得到两种或三种以上的金属阳离子溶液,然后将多级孔结构的无机固体酸在超声搅拌条件下加入到上述溶液中,超声方法可以是bath超声或Tip超声,超声功率为1-20W/(ml样品),频率40KHz-80MHz,沉淀温度2℃-60℃,时间1h-12h,搅拌速率50-500rpm,再经过洗涤、干燥、焙烧,洗涤方法可以是过滤洗涤或离心洗涤,使洗涤后pH在6.5-7.5,干燥温度是80-150℃,时间大于12h,焙烧温度是300-650℃,时间1-3h;
所述物理复合中的球磨法,温度为25~100℃,载气为:a)氮气和/或惰性气体,b)氢气与氮气和/或惰性气体的混合气,其中氢气于混合气中的体积为5~50%,c)CO与氮气和/或惰性气体的混合气,其中CO于混合气中的体积为5~20%,或d)O2与氮气和/或惰性气体的混合气,其中O2于混合气中的体积为5-20%,所述惰性气体为氦气、氩气、氖气中的一种或两种以上;
所述物理复合中的机械混匀法,是指使用包括3D旋转混匀仪、行星式混匀仪、或涡轮混匀仪等混匀装置,将多组分金属复合物和多级孔结构的无机固体酸加入容器中,利用这些物料及容器的高速运动产生的挤压力、撞击力、裁剪力、摩擦力等中的一种或两种以上作用实现分离、破碎、混匀等目的,通过调变温度与载气气氛实现机械能、热能与化学能的转换,进一步增强不同组分间的相互作用;反应过程中,设置温度20-100℃,载气气氛为:a)氮气和/或惰性气体,b)氢气与氮气和/或惰性气体的混合气,其中氢气于混合气中的体积为5~50%,c)CO与氮气和/或惰性气体的混合气,其中CO于混合气中的体积为5~20%,d)O2与氮气和/或惰性气体的混合气,其中O2于混合气中的体积为5-20%,所述惰性气体为氦气、氩气、氖气中的一种或两种以上。
使用任一所述催化剂进行催化反应,在所需反应温度、反应压力条件下,反 应原料气体H2/CO体积比=0.5/1~4/1,制备C2-C4低碳烯烃,即含有两个碳原子至四个碳原子数的烯烃,包括乙烯、丙烯和丁烯中的一种或者一种以上。
所述催化反应,进行该反应前,包括一个催化剂预处理过程,预处理气氛为a)H2和CO的混合气,其中H2/CO体积比为0.5/1~4/1;
或,b)氢气与惰性气体的混合气;b)中,氢气在混合气中的体积百分比为5~100%,惰性气体为氮气、氦气、氩气或氖气中的一种或两种以上;
或,c)CO与惰性气体的混合气;c)中,CO在混合气中的体积百分比为5~100%,惰性气体为氮气、氦气、氩气或氖气中的一种或两种以上;
所述催化剂预处理的温度为250~600℃,预处理压力为0.1~3.0MPa,预处理的空速为500~5000h-1,空速优选为1000~4000h-1;
所述金属复合物经过含氢气或一氧化碳的气氛还原活化之后,表面结构中具有氧空位,即氧为配位不饱和状态,可高效地催化活化CO生成中间态物种CHx(其中x=1,2或3)中的一种或两种以上的物种,表面氧物种与CO反应生成CO2,这些极其活泼的CHx物种与CO结合生成CHxCO,生成的中间态物种在表面为弱吸附,极易从表面脱附并扩散至无机固体酸的孔道内,被进一步催化转化反应生成低碳烃,无机固体酸的孔道大小和酸性可以调变低碳烃的选择性以及其中烯烃和烷烃的相对比例。
所述反应原料气体为混合气,混合气中含有H2、CO、以及其他气体,其他气体为惰性气体和/或非惰性气体中的一种或者两种;
所述惰性气体为氮气、氦气、氩气、氖气中的一种或两种以上,在反应原料气体中的体积含量小于10%;
所述非惰性气体为二氧化碳、气化的水、甲醇、乙醇中的一种或者两种,在反应原料气体中的体积含量小于50%。
H2与CO的体积比H2/CO为0.5/1~4/1。
所述反应采用的装置为流化床、移动床或固定床;
所述反应温度为300~500℃,反应温度优选为350~450℃,空速为500~10000h-1,空速优选为2000~7000h-1;
所述反应为连续流动反应模式或间歇反应模式;
所述连续流动反应模式中,反应压力为0.1~6.0MPa,反应压力优选为1.0~5.0MPa;
所述间歇反应模式中,反应压力为1.0~10MPa,反应压力优选为1.0~5.0MPa。
表1无机酸盐的阳离子和阴离子列表
本发明涉及一种由合成气一步法直接转化制低碳烯烃的方法,低碳烯烃产物包括乙烯、丙烯和丁烯中的两种或者两种以上;低碳烯烃产物在所有碳氢化合物中含量为50~85%。
本发明的有益效果:
本发明与传统费托反应路径一步法制低碳烯烃的过程相比,具有如下特点:
1.产物不同于由链增长控制的Anderson Schultz Flory分布规律;传统费托反应产物中,可预测的C2-C4烃选择性最高为58%,而本发明提供产物中C2-C4的选择性可达60~95%;
2.低碳烯烃在总烃产物中的选择性高达50~85%;
3.产物中甲烷选择性和C5+长碳链碳氢化合物的选择性极低,两者共为5~10%;
4.催化剂不同于传统费托反应制低碳烃所用的过渡金属如Fe、Co等为主成分的催化剂,而是含多级孔结构的无机固体酸与多组分金属复合物的复合催化材料。
本发明与目前已经工业化的两段法,即先将合成气转化成甲醇或二甲醚,再将甲醇或二甲醚转化成低碳烯烃的过程相比,具有如下特点:
1.一个反应器,一个催化剂,一步法转化,可显著降低设备投资和生产成本;
2.催化剂为含多级孔结构的无机固体酸与多组分金属复合物复合而成的催化材料,产品选择性可以很好调控;
3.该催化剂稳定性好,寿命长。
4.本发明具有操作单元单一,过程重复性好,产物选择性高,催化剂寿命长(>500h),具有广阔的工业应用前景。
本发明所用的催化剂具有如下优点:在合成气制备低碳烯烃反应中,本发明提供的催化剂为多组分金属复合物与多级孔结构的无机固体酸复合得到的复合材料。该催化剂不同于传统费托反应制低碳烃所用的过渡金属为主成分的催化剂,也不同于工业两段法经过甲醇合成所用的催化剂,其创新之处包括结构和功能:结构方面在于催化剂同时具有微孔、介孔的多级孔结构,这种独特结构使催化剂具有显著的优势,即在反应条件下长时间运行(>500h)而不失活;功能方面在于可以直接催化合成气转化,无需配套高能耗的水煤气变换过程,无需将合成气转化成甲醇并提取甲醇的过程,也不同于传统的链增长机理,一步可以生成低碳烃产物,其选择性为60~95%,明显突破了传统费托反应中的Anderson-Schultz-Flory分布限制,且甲烷和长碳链产物(C5+)的选择性低于5~10%。
附图说明
图1实施例8中核壳结构的Cr1Zn1@SiAl的SEM图;
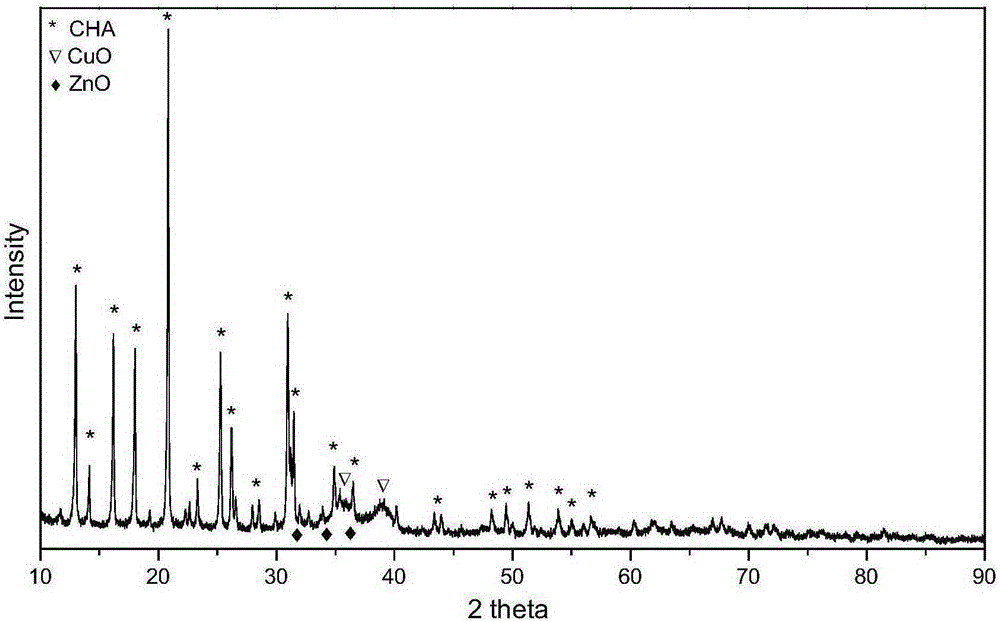
图2实施例8中Cu1Zn1@SiAl的XRD图;
图3实施例14中多级孔结构的硅磷铝无机固体酸的N2物理吸附曲线及其孔径分布图;
图4实施例6中合成气直接转化为产物的在线色谱分析的色谱图。
具体实施方式
下面通过实施例对本发明做进一步阐述,但是本发明的权利要求范围不受这些实施例的限制。同时,实施例只是给出了实现此目的的部分条件,但并不意味着必须满足这些条件才可以达到此目的。
催化反应实例
以连续流动固定床反应为例,但是催化剂也适用于流化床反应器。该装置配备气体质量流量计、气体脱氧脱水管和在线产物分析色谱(反应器的尾气直接与色谱的定量阀连接,进行周期实时采样分析)。上述催化剂在使用前,均压片过筛至20-40目颗粒或者40-60目,备用。
将下述实施例或对比例中所述的催化剂2.8g,置于固定床反应器中,使用Ar置换反应器中的空气,然后在纯H2气氛中310~350℃下还原1h,在Ar气氛中降至室温,再切换合成气(5%Ar;H2/CO=2/1),升温至反应温度,调节反应原料气的空速、压力。产物由在线色谱检测分析。改变温度、压力、空速和H2/CO比例,可以改变反应性能。
实施例1
化学复合法制备多组分金属复合物-多级孔结构的硅铝无机固体酸复合催化剂。
以CuZnAl多组分金属复合物,多级孔结构的硅铝无机固体酸为例。
按氧化物SiO:Al2O3:Na2O:R2O:H2O=40:1:16:5:900(质量比)称取原料:硅溶胶30%(质量浓度);硫酸铝;氢氧化钠;N,N,N-一三甲基金刚烷氢氧化铵(R);去离子水,室温混合后,在60℃,200rpm速度搅拌24h后转移到水热釜中,155℃下晶化6天。自然冷却到室温,转移到烧杯中水浴70℃,按100ml原液:4g氯化铵的比例加入氯化铵,恒温搅拌3h,反复离心洗涤使得洗涤结束时上清液pH是7,沉淀物于120℃下烘干24h后,在650℃空气中焙烧3h得到多级孔结构的硅铝无机固体酸。
称取制备好的多级孔结构的硅铝无机固体酸5.3g,取三水合硝酸铜1.2g,六水合硝酸锌1.49g,九水合硝酸铝1.8g,溶解于100ml水溶液中,通过浸渍法将CuZnAl引入到多级孔结构的硅铝无机固体酸上,并经过室温真空干燥,500℃静态空气中焙烧1h,得到元素摩尔比为1:1:1的Cu1Zn1Al1/mesoSiAl,多组分金属复合物的负载量为20wt%。根据此方法,可以改变各金属组分及其比例,以及多组分金属复合物的负载量。
氮气物理吸脱附表征显示该催化剂具有多级孔结构,微孔比表面为347m2/g,微孔孔径分布在介孔比表面积为214m2/g,大孔比表面为62m2/g;介孔孔径大小分布在2-15nm。X射线衍射(XRD)表征显示,多组分金属复合物含CuO、ZnO、Al2O3、ZnAlO2和具有CHA结构的硅铝无机固体酸,金属氧化物的晶粒大小5-15nm;无机固体酸晶粒大小为40-70nm;扫描电镜(SEM)表征表明,多级孔结构的硅铝无机固体酸二次粒子大小为10-100μm,无机固体酸晶粒大小为10-100nm;高分辨透射电镜(HRTEM)表征表明,多组分金属复合物的晶粒大小5-15nm,二次粒子大小15-50nm;中强酸对应的NH3脱附峰温度为360℃,中强酸位点的量是2mol/kg。中强酸是按照NH3-TPD定义的,是根据NH3的脱附峰位置,所述脱附峰的位置是指在标准测试条件下,在样品质量w与载气流速f比值(w/f)=100g·h/L,10℃/min升温速度的测试条件下,TCD记录脱附NH3的热导信号,绘制脱附曲线,根据曲线峰位置顶点将所述无机固体分为三种酸性强度:所述中强酸是NH3脱附温度在275-500℃的酸性位。
实施例2
制备过程与实施例1基本相同,不同之处制备实施例1中多级孔结构的硅铝无机固体酸的制备过程中,改变晶化温度为150℃,5天,制备出中强酸位点的量是0.8mol/kg的CHA结构的硅铝无机固体酸。
作为对比例1,制备过程与实施例1基本相同,不同之处制备实施例1中多级孔结构的硅铝无机固体酸的制备过程中,晶化温度为140℃,3天,制备出中强酸位点的量是0.01mol/kg的硅铝无机固体酸。
作为对比例2,制备过程与实施例1基本相同,不同之处制备实施例1中多级孔结构的硅铝无机固体酸的制备过程中,晶化温度为140℃,22h,制备出中强酸位点的量是0.005mol/kg的硅铝无机固体酸。
在365℃,2MPa体积空速4000h-1的反应条件下,反应结果如下表。
对比例1,2中转化率极低,这是由于固体酸中中强酸位较少,导致多组分金属复合物上产生的中间物种不能被及时、很好地转化成为目标产物。同时甲烷 选择性大大提高,超过30%,而低碳烯烃选择性也降低的很明显。相比之下,含有较多中强酸的本发明催化剂,转化率大大提高,而且目标产物烯烃的选择性也高。实施例1转化率明显高于实施例2的转化率因为该催化剂是优选范围内。可见多级孔结构的硅铝无机固体酸的酸性含量对催化性能包括转化率和选择性的调控极其重要。
实施例3
化学复合法制备多组分金属复合物-多级孔结构的硅磷铝无机固体酸复合催化剂
按氧化物SiO:Al2O3:H3PO4:R:H2O=18:16:32:55:150(质量比)称取原料:硅溶胶30%(质量浓度);硫酸铝;磷酸;TEA(R);去离子水,室温混合后,在30℃下搅拌老化,500rpm速度搅拌72h后转移到水热釜中,220℃下晶化15h。水浴骤冷到室温,反复离心洗涤使得洗涤结束时上清液pH是7,沉淀物于130℃下烘干17h后,在580℃空气中焙烧5h得到多级孔结构的硅磷铝无机固体酸。
称取所述多级孔结构的硅磷铝无机固体酸2.1g,取六水合硝酸锌1.54g、九水合硝酸铬3.8g,溶解于水溶液中配成混合液A,称取6.3g碳酸铵,溶解于100ml水中配成溶液B,50℃下将溶液B逐滴加入以7W功率tip超声,400rpm速度搅拌混合液A中,离心洗涤使得洗涤结束时上清液pH是7并经过110℃空气中干燥,500℃静态空气中焙烧2h,得到ZnCr1.8/SiPAl。然后,以此为载体,准确称取0.057g醋酸钯Pd(Ac)2溶解在丙酮中,通过浸渍引入到ZnCr1.8/SiPAl上,其中Pd与Zn的摩尔比为0.01,得到元素摩尔比为0.01:1:1.8的Pd0.01ZnCr1.8/SiPAl,多组分金属复合物的负载量为20wt%。根据此方法,可以改变各金属组分及其比例,以及多元金属复合物的负载量。
氮气物理吸脱附表征显示该催化剂具有多级孔结构,微孔孔径分布在介孔孔径大小分布在2-8nm,总的比表面积为416m2/g,其中介孔比表面积占总比表面积的35%,大孔比表面占总面积20%,X射线衍射(XRD)表征显示,多组分金属复合物含Cr2O3、CrO3、ZnO、Al2O3、ZnCr2O4,和CHA结构的硅铝无机固体酸,金属氧化物晶粒大小7-16nm,无机固体酸晶粒大小为50-60nm;扫描电镜(SEM)表征表明,硅铝无机固体酸二次粒子大小为100-500μm;高分辨透射电镜(HRTEM)表征表明,多组分金属复合物的晶粒大小7-16nm,二次粒子大小20-100nm;中强酸对应的NH3脱附峰温度为386℃,中强酸位点的量是6mol/kg。
实施例4
制备过程与实施例3基本相同,不同之处将硅铝无机固体酸制备时,改变搅拌时间改变为3h,晶化时间变为24h,制备出总比表面积为377m2/g,介孔比表面积占总比表面积的21%,大孔比表面积占总比表面积的20%的CHA结构硅铝无机固体酸;中强酸对应的NH3脱附峰温度为385℃,中强酸位点的量是5.8mol/kg。
作为对比例3,制备过程与实施例3基本相同,不同之处在于制备硅铝无机固体酸时,老化温度改为80℃,搅拌2天后,在240℃下晶化24h,获得总比表面积为232m2/g,介孔比表面积占总比表面积的4%,大孔比表面积占总比表面积的1%,的CHA结构硅铝无机固体酸;中强酸对应的NH3脱附峰温度为411℃,中强酸位点的量是2mol/kg。
作为对比例4,制备过程与实施例3基本相同,不同之处在于制备硅铝无机 固体酸时,老化温度改为30℃,搅拌2天后,在240℃下晶化48h,获得总比表面积为287m2/g,介孔比表面积占总比表面积的10%,大孔比表面积占总比表面积的4%的CHA结构硅铝无机固体酸;中强酸对应的NH3脱附峰温度为390℃,中强酸位点的量是5mol/kg。
作为对比例5,制备过程与对比例3基本相同,不同之处在于制备硅铝无机固体酸时,按照比表面积计算,利用介孔分布在4-8nm的商品SBA-15分子筛与对比例3中硅铝无机固体酸机械混合,混合比例按照硅铝无机固体酸与SBA-15比表面积为13:7,然后进行研磨混合,得到具有介孔与微孔复合物的硅铝无机固体酸,其中微孔占总比表面积的62%,介孔比表面积占总比表面积的36%,大孔占总比表面积的2%。但直径2nm-50nm的介孔孔道不是由无机固体酸晶粒堆积而成,其中一级孔不位于相邻的二级孔和/或三级孔孔壁,而是一级孔自己孤立存在,而二级三级孔道孤立存在,不符合权利要求所述的三种孔道之间相互连接贯通,未构成三维多级孔道结构。
在400℃,2.5MPa体积空速4000h-1的反应条件下,反应结果如下表。
实施例3中低碳烯烃的选择性明显优于对比3、4中的选择性,而实施例3中的选择性比实施例4中的更优,这是由于实施例3催化剂的中硅铝无机固体酸介孔比表面积占总比表面积的35%,大孔比表面占总面积20%,相比实施例4中介孔比表面积占总比表面积的21%,大孔比表面积占总比表面积的20%,在更加优选的范围内,所以传质更优,抑制了加氢产生烷烃,选择性大幅提高。而对比例中低碳烯烃的选择性远低于本发明的催化剂,这是由于分子筛中介孔及大孔过少,没有达到权利要求所述的介孔与大孔的含量,不利于传质,同时酸强度的变化也使得产物过度加氢产生烷烃,大大降低了低碳烯烃的选择性。而对比例5中虽然介孔与大孔的含量复合权利要求所述的范围,但是其含有的三种孔道直接没有相互连接贯通,未构成权利要求所述的三维三级孔道结构,另外SBA-15本身表面及其孔道内,中强酸很少,所以对比例5中的中强酸主要分布在微孔中,而不是均匀分布在所述的三维三级孔道内不符合权利要求5所述,所以与对比例3相比没有实质性的改变,低碳烯烃的选择性仍然很低。相比之下,介孔比表面积占20-75%,大孔占20-65%构成三维多级孔道结构的催化剂上,烯烃的选择性超过60%,不仅远高于对比例3,4,也高于传统费托合成的低碳烯烃的理论选择性58%,可见无机酸的孔道结构对产物选择性的调控极其重要。
实施例5
物理复合法制备多组分金属复合物-多级孔结构的硅磷铝无机固体酸复合催化剂该方法包括通过球磨,机械混匀仪,摇床,振荡混合等机械化学方式对不同材料进行混合的方法。
多级孔结构的硅磷铝无机固体酸:以三乙胺、明胶与戊二醛的交联物作为模板。将10g异丙醇铝、5.30g浓磷酸、3.67g硅溶胶(含30%质量浓度)、1.46g三乙胺、13.2g H2O混合,于室温搅拌3h,搅拌速度400rpm,然后加入1.68g 2wt%的明胶溶液,升温到57℃,继续搅拌0.5h。将得到的混合液倒入培养皿中,于0℃冷冻12h,得到冻胶,加入适量的5wt%戊二醛进行交联反应,反应后的悬浮液于10000rpm离心,得到的固体样品置于50℃烘箱中干燥过夜后,装入聚四氟乙烯内衬中,再将此小内衬放入大号聚四氟乙烯内衬中,大号内衬中需预先加入适量三乙胺与水的混合液,然后大小内衬一起装入不锈钢自压晶化釜中,于200℃干胶法晶化36h,反复离心洗涤使得洗涤结束时上清液pH是7,沉淀物于90℃下烘6h,在110℃下烘干12h后,在550℃空气中焙烧3h,得到多级孔结构的硅磷铝无机固体酸。
按照金属元素Zn:Cu:Co:Cr:Al摩尔比为6:0.2:1:1:1的比例分别称取六水合硝酸锌17.33g,六水合硝酸铜,六水硝酸钴,九水合硝酸铬,溶于100ml去离子水中,加入γ-Al2O3,于25℃下超声搅拌,tip超声功率5W,搅拌速率300rpm,然后110℃空气中干燥,500℃焙烧,得到元素摩尔比为6:0.2:1:1:1的多组分金属复合物Zn6Cu0.2Co1Cr1Al1。取该多组分金属复合物2g,多级孔结构的硅磷铝无机固体酸5g,置于聚四氟乙烯混匀罐中,首先用氦气将其中的空气置换3次,通入2%H2(He平衡)之后闭上混匀罐,开始混匀处理,速率为450转/分钟,5分钟后结束,制得复合催化剂Zn6Cu0.2Co1Cr1Al1/SiPAl,其中多组分金属复合物占催化剂的总质量40wt%。依此方法,可以改变多组分金属复合物的含量和多级孔结构的无机固体酸的种类。
氮气物理吸脱附表征显示该催化剂具有多级孔结构,微孔比表面为243m2/g,微孔孔径大小分布在介孔比表面积为318m2/g,大孔比表面为182m2/g,介孔孔径大小分布在15-25nm。中强酸对应的NH3脱附峰温度为367℃,中强酸位点的量是1.5mol/kg。
实施例6,制备过程与实施例5基本相同,不同之处在于按催化剂的总质量为100%计,将多组分金属复合物的含量调为15wt.%。
作为对比例6,制备过程与实施例5基本相同,不同之处在于按催化剂的总质量为100%计,将多组分金属复合物的含量调为5wt.%。
作为对比例7,制备过程与实施例5基本相同,不同之处在于按催化剂的总质量为100%计,将多组分金属复合物的含量调为90wt.%。
实施例7
制备过程与实施例5基本相同,不同之处在于多级孔结构的硅磷铝无机固体酸制备中更改晶化温度为190℃,时间为33h,焙烧温度600℃,时间3h。氮气物理吸脱附表征显示该催化剂具有多级孔结构,介孔孔径大小分布在10-15nm。扫描电镜(SEM)表征表面,硅铝无机固体酸二次粒子大小为10-70μm,无机固体酸晶粒大小为60-80nm;中强酸对应的NH3脱附峰温度为385℃,中强酸位点的量是1.5mol/kg。
作为对比例8,制备过程与实施例7基本相同,不同之处在于多级孔结构的硅磷铝无机固体酸制备中更改晶化温度为160℃,时间为24h,焙烧温度550℃,时间8h,流动空气下进行焙烧。氮气物理吸脱附表征显示该催化剂具有多级孔 结构,介孔孔径大小分布在2-5nm。扫描电镜(SEM)表征表面,硅铝无机固体酸二次粒子大小为5-100nm;XRD显示无机固体酸晶粒大小为1-5nm;中强酸对应的NH3脱附峰温度为349℃,中强酸位点的量是0.06mol/kg。
在370℃,2MPa体积空速6000h-1的反应条件下,反应结果如下表
对比例6,7中转化率远远低于实施例中的转化率,且对比例6中的甲烷选择性高达70%,而对比例7中的碳5以上的选择性超过40%,低碳烯烃的选择性也都低于本发明催化剂的结果,这是由于对比例的多组分金属复合物在整个催化剂中的含量不符合权利要求书所述的含量范围,不利于反应过程中生成的中间物种的转化。而实施例5与实施例6比较,二者的低碳烯烃的选择性都超过了50%,且甲烷选择性都低于5%,但是实施例5的转化率更高,这是因为实施例5中多组分金属复合物在整个催化剂中的含量在优选范围,所以,转化率更高。
实施例6,7中多级孔结构的硅磷铝无机固体酸的晶粒大小及其堆积成的二次粒子大小及介孔孔道大小都复合权利要求的范围,但相对于实施例6,实施例7的晶粒大小,二次粒子大小及介孔孔道在优选范围,所以表现出了更好的转化率及选择性,而对比例8中因为不符合权利要求的范围,表现为无机固体酸结晶程度不佳,进而不能很好的进行催化转化,所以转化率,选择性都明显要差很多。所以无机固体酸的晶粒尺寸及二次粒子尺寸以及介孔孔道的尺寸都对催化转化及产物选择性的调控至关重要。
实施例8
包覆生长法制备多组分金属复合物-多级孔结构的无机固体酸复合催化剂。
该方法涉及在制得的多元金属复合物上,通过水热等方式生长出一层或部分的多级孔结构的无机固体酸材料得到。
以CuZn金属和共沉淀法为例,方法不限于双金属,且适用于其它金属,并可以改变不同金属的相对比例,可用氨水、碳酸铵或碳酸氢铵作为沉淀剂。
称取三水合硝酸铜4.8g,六水合硝酸锌5.95g溶于100ml去离子水中。称取碳酸铵11g溶于去离子水100ml中。两组溶液同时滴加到70℃恒温超声搅拌的去离子水中共沉淀,超声功率是5W,搅拌速率500rpm。然后维持70℃老化3h,用70℃去离子水洗涤到中性。然后,在110℃下干燥12h,经过500℃下焙烧1h,制备得到CuZn复合氧化物,其中Cu/Zn摩尔比为1/1。依此方法, 可得到不同摩尔比的CuZn氧化物。
取10ml硅溶胶溶液(含30wt%SiO2),加入10ml去离子水中,称取1g上述多组分金属复合物加入其中,摇床浸润2h,分离固体液体,放入120℃烘箱干燥12h,在马弗炉中500℃下焙烧2h,使上述氧化物外面覆盖一层SiO2膜,进一步取11.37g TPAOH,9.68g乙醇,0.06g三氧化二铝,50ml H2O搅拌溶解。加入上述氧化物,搅拌,称取9.65g TEOS滴加至上述溶液中,搅拌6h。在180℃下晶化72h,过滤洗涤,60℃烘箱干燥,500℃下焙烧5h,得到具有核壳结构的Cu1Zn1@SiAl催化剂,其中氧化物占催化剂总质量为50wt%。根据此方法,可以改变各金属组分及其比例,以及多元金属复合物的负载量。
氮气物理吸脱附表征显示该催化剂具有多级孔结构,微孔比表面为311m2/g,微孔孔径大小分布在介孔比表面积为203m2/g,与大孔比表面为171m2/g,介孔孔径大小分布在13-30nm。X射线衍射(XRD)表征显示,多组分金属复合物含CuO、ZnO和呈MFI结构的硅铝无机固体酸,金属氧化物晶粒大小8-20nm,无机固体酸晶粒大小为55-70nm;扫描电镜(SEM)观察硅铝无机固体酸二次粒子大小为30-60μm;高分辨透射电镜(HRTEM)表征表明,多组分金属复合物的晶粒大小8-20nm,二次粒子大小20-100nm;中强酸对应的NH3脱附峰温度为408℃,中强酸位点的量是0.7mol/kg。
实施例9
制备过程和方法与实施例8基本相同,不同之处在于多元金属复合物的制备过程中,更换前驱物为六水合硝酸锌,六水合硝酸钴,九水合硝酸铝,其中六水合硝酸锌质量是5.95g,按照元素摩尔比为Zn:Co:Al=1:0.01:1称量前驱体溶于100ml去离子水中。称取碳酸铵14.3g溶于去离子水100ml中。
氮气物理吸脱附表征显示该催化剂具有多级孔结构,微孔比表面为297m2/g,微孔孔径分布在介孔比表面积为122m2/g,大孔比表面为111m2/g,介孔孔径大小分布在2-8nm。X射线衍射(XRD)表征显示,多组分金属复合物含Co3O4、Co2O3、ZnO、Al2O3、ZnCo2O4和CHA结构的硅铝无机固体酸,金属氧化物晶粒大小9-17nm,无机固体酸晶粒大小为50-60nm;扫描电镜(SEM)表征表明,硅铝无机固体酸二次粒子大小为100-500μm;高分辨透射电镜(HRTEM)表征表明,多组分金属复合物的晶粒大小9-17nm,二次粒子大小20-100nm;中强酸对应的NH3脱附峰温度为363℃,中强酸位点的量是0.6mol/kg。
作为对比例9,制备过程与实施例8基本相同,不同之处在于合成多元金属复合物过程中,不使用超声处理,在90℃老化0.5h,且焙烧温度改为300℃,使得最终催化剂不能形成很好的晶型,导致最后获得的晶粒大小<1nm。在包覆无机固体酸的过程中控制焙烧时间为1.5h。最终催化剂的微孔比表面为148m2/g,微孔孔径大小分布在介孔比表面积为27m2/g,大孔比表面为7m2/g,介孔孔径大小分布在20-47nm。金属氧化物晶粒大小<1nm,二次粒子大小10nm-50μm;无机固体酸晶粒大小为53-79nm;无机固体酸二次粒子大小为300-600μm;中强酸对应的NH3脱附峰温度为410℃,中强酸位点的量是0.05mol/kg。
作为对比例10,制备过程与实施例8基本相同,不同之处在于合成多元金属复合物过程中,超声处理功率0.5W,在30℃老化3天,且焙烧温度改为800℃, 使得最终催化剂的晶粒大小>30nm。最终催化剂的微孔比表面为757m2/g,微孔孔径大小分布在介孔比表面积为147m2/g,大孔比表面为150m2/g,介孔孔径大小分布在8-14nm。金属氧化物晶粒大小30nm-70nm,二次粒子大小100nm-50μm;无机固体酸晶粒大小为80-120nm;无机固体酸二次粒子大小为200-800μm;中强酸对应的NH3脱附峰温度为430℃,中强酸位点的量是4mol/kg。
实施例8、对比例9和对比例10,在370℃,2MPa体积空速7000h-1的反应条件下,反应结果如下表。
实施例9在400℃,2.5MPa体积空速4000h-1的反应条件下,反应结果如下表。
对比例9多元金属复合物的晶粒大小都非常小,其中很多金属甚至以单原子或者少量原子形成的原子簇形式存在,粒子大小小于权利要求所述的优选范围也小于权利要求所述的晶粒大小范围,对比例9反应结果虽然转化率有所增加,但甲烷选择性大大增加超过30%,同时低碳烯烃选择性下降到不足20%。而对比例10中,多元金属复合物的晶粒大小大于权利要求所述的晶粒大小范围上限,粒子过大,比表面积降低,导致其活化转化合成气能力不足,转化率非常低,同时加氢比较严重,低碳烯烃的选择性也有所下降。所以金属复合物的晶粒大小对于调控催化性能至关重要。
实施例10
制备过程和方法与实施例5基本相同,不同之处在于多元金属复合物的制备过程为,分别称取六水合硝酸锌17.33g,六水硝酸钴0.17g,氯化钛22.1g,溶于300ml去离子水中,称取碳酸铵60.5g溶于去离子水300ml中。两组溶液同时滴加到70℃恒温超声搅拌的去离子水中共沉淀,超声功率是5W,搅拌速率500rpm。然后维持70℃老化3h,用70℃去离子水洗涤到中性。然后,在110℃下干燥12h,经过500℃下焙烧1h,制备得到ZnCo0.05Ti2复合氧化物,其中Co/Ti/Zn元素摩尔比为0.05/2/1。依此方法,可得到不同摩尔比的ZnCoTi氧化物。
氮气物理吸脱附表征显示该催化剂具有多级孔结构,微孔比表面为251m2/g,微孔孔径大小分布在介孔比表面积为213m2/g,大孔比表面为216m2/g,介孔孔径大小分布在4-13nm。X射线衍射(XRD)表征显示,多组分金属复合物含ZnO、Co3O4、TiO2、ZnCo2O4和呈CHA结构的硅铝无机固体酸,金属氧化物的晶粒大小8-20nm,无机固体酸晶粒大小为60-80nm;扫描电镜(SEM)表征表面,硅铝无机固体酸二次粒子大小为300-700μm;高分辨透射电镜(HRTEM)表征表明,多组分金属复合物的晶粒大小8-20nm,二次粒子大小30-120nm;中强酸对应的NH3脱附峰温度为377℃,中强酸位点的量是2mol/kg。
作为对比例11,制备过程与实施例10基本相同,不同之处在于称量金属前驱体时,改变多元金属复合物中金属元素的比例为Zn0.05Co3Ti2,按照此比例配置金属前驱体溶液进行合成。XRD表征显示,多组分金属复合物含有ZnO、CoO、Co、ZnCo2O4、CoTiO3,且按原子数量计,金属Co含量在所有金属原子中达到30%,超过了权利要求所述的10%。多组分金属复合物的晶粒大小8-40nm,二次粒子大小30-120nm。
在400℃,2MPa体积空速6000h-1的反应条件下,反应结果如下表。
对比例11,在相同条件下反应产物中碳5以上产物超过50%,且甲烷也超过20%,因为其中金属态的Co含量过高,不在权利要求的范围内,使得反应趋向于常规的费托合成路径,产物受到ASF分布的限制,导致低碳烯烃选择性低,而生成了较多长碳链的碳氢化合物。
实施例11
制备过程和方法与实施例8基本相同,不同之处在于得到的多级孔结构的硅磷铝无机固体酸,按照质量比1:500至于10%硝酸铵溶液中80℃搅拌8h后,抽滤并在室温干燥后,再按照质量比1:500至于1%硝酸铜水溶液中80℃搅拌8h后,抽滤并在室温干燥后,置于110℃烘箱干燥14h后,500℃空气焙烧1h得到Cu离子交换后的多元金属复合物。
多组分金属复合物的制备过程差别在于,金属前驱体的配制过程是称取六水合硝酸锌5.95g,按照元素摩尔比为1:1称取钼酸铵,溶于100ml去离子水中。称取碳酸铵11g溶于去离子水100ml中。
氮气物理吸脱附表征显示该催化剂具有多级孔结构,微孔比表面为239m2/g,微孔孔径大小分布在介孔比表面积为271m2/g,与大孔比表面为198m2/g,介孔孔径大小分布在4-13nm。X射线衍射(XRD)表征显示,多组分金属复合物含ZnO、CuO、MoO3、ZnMoO4和呈CHA结构的硅铝无机固体酸,金属氧化物的晶粒大小12-26nm,无机固体酸晶粒大小为60-90nm;扫描电镜(SEM)表征表面,硅铝无机固体酸二次粒子大小为300-700μm;高分辨透射电镜(HRTEM)表征表明,多组分金属复合物的晶粒大小12-26nm,二次粒子大小30-120nm;中强酸对应的NH3脱附峰温度为345℃,中强酸位点的量是0.5mol/kg。
实施例12
制备过程与实施例11基本一致,不同之处在于更换了多元金属复合物中的金属元素,保证替换金属的与被替换金属的原子数量相等,在配置金属前驱体溶液时将钼酸铵替换成硝酸钒酰。
实施例13
制备过程与实施例11基本一致,不同之处在于更换了多元金属复合物中的金属元素,保证替换金属的与被替换金属的原子数量相等,在配置金属前驱体溶液时将钼酸铵替换成硝酸锰。
在360℃,0.8Mpa体积空速3000h-1的反应条件下,反应结果如下表。
实施例11与实施例12,13差别在于更换了多元金属复合物中的金属元素组成,从Mo到V到Mn。随之低碳烯烃的选择性逐渐升高。这与权利要求所述的金属元素优选所一致。
实施例14
制备过程和方法与实施例3基本相同,不同之处在于
多级孔结构的硅磷铝无机固体酸制备过程和方法是,按照质量比SiO2:Al2O3:R:H2O=300:15:30:100称取水玻璃,薄水铝石,乙二胺,水于250ml烧杯中50摄氏度搅拌老化1h后,在30℃下搅拌二次老化,500rpm速度搅拌72h后转移到水热釜中150℃烘箱中晶化47h,水浴骤冷到室温,反复离心洗涤使得洗涤结束时上清液pH是7,沉淀物于110℃下烘干17h后,在550℃空气中焙烧3h得到多级孔结构的硅铝无机固体酸。
多组分金属复合物其金属前驱体的配制过程是称取六水合硝酸锌5.95g,按照元素摩尔比为Zn:Pd:Zr=1:0.01:1称取硝酸钯,五水合硝酸锆,溶于100ml去离子水中。称取碳酸铵11.3g溶于去离子水100ml中。
氮气物理吸脱附表征显示该催化剂具有多级孔结构,微孔比表面为239m2/g,微孔孔径大小分布在介孔比表面积为308m2/g,与大孔比表面为161m2/g,介孔孔径大小分布在4-13nm。X射线衍射(XRD)表征显示,多组分金属复合物含ZnO、PdO、ZrO2、ZnZr2O4和呈CHA结构的硅铝无机固体酸,金属氧化物的晶粒大小12-22nm,无机固体酸晶粒大小为60-90nm;扫描电镜(SEM)表征表面,硅铝无机固体酸二次粒子大小为300-700μm;高分辨透射电镜(HRTEM)表征表明,多组分金属复合物的晶粒大小12-22nm,二次粒子大小40-120nm;中强酸对应的NH3脱附峰温度为370℃,中强酸位点的量是1.6mol/kg。
实施例15
制备过程与实施例14基本一致,不同之处在于在配置金属前驱体溶液时改变了多元金属复合物的金属前驱体的配制过程的金属元素Zn与其他元素的原子比例为5:1。
作为对比例12,制备过程与实施例14基本一致,不同之处在于在配置金属前驱体溶液时改变了多元金属复合物的金属前驱体的配制过程中金属元素Zn与其他元素的原子比例为0.05:1。
在380℃,1.5MPa体积空速8000h-1的反应条件下,反应结果如下表。
实施例14与对比例12改变了Zn与其他元素的比例,对比例12的比例不在权利要求的比例范围,所以CO的转化率很低,而实施例15虽然比例在权利要求范围内但是不在优选范围内所以转化率也较低。
实施例16
多级孔结构的硅磷铝无机固体酸制备过程和方法与实施例3基本相同,不同之处在于将得到的多级孔的硅磷铝无机固体酸置于安瓶接入10-2pa以上的真空恒温区,对多级孔的硅磷铝无机固体酸进行加热,5℃/min升温到420℃恒温12h处理后,密封转移到Ar气氛的手套箱中,将多级孔的硅磷铝无机固体酸与氯化钼按照质量比1:0.1置于石英安瓶两侧,再次接入10-2pa真空后,对石英安瓶进行封管处理使得多级孔的硅磷铝无机固体酸与氯化钼同时被封入真空的石英安瓶内部。将内封有多级孔的硅磷铝无机固体酸与氯化钼的安瓶置于管式炉中,以10℃/min升温到400℃恒温24h后,降到室温,打破安瓶得到氧化钼载量为1%的Mo-SiAlP。
多组分金属复合物的制备过程及二者复合方式与实施例5基本一致,不同之处在于金属前驱体的配制过程是,称取六水合硝酸锌5.96g,按照元素摩尔比Zn:Al=1:1称取九水合硝酸铝,溶于100ml去离子水中。
氮气物理吸脱附表征显示该催化剂具有多级孔结构,微孔比表面为533m2/g,微孔孔径分布在介孔比表面积为133m2/g,大孔比表面为118m2/g,介孔孔径大小分布在3-8nm。X射线衍射(XRD)表征显示,多组分金属复合物含MoO2、ZnO、Al2O3、ZnAl2O4和CHA结构的硅铝无机固体酸,金属氧化物晶粒大小7-12nm,无机固体酸晶粒大小为50-60nm;扫描电镜(SEM)表征表明,硅铝无机固体酸二次粒子大小为100-500μm;高分辨透射电镜(HRTEM)表征表明,多组分金属复合物的晶粒大小7-12nm,二次粒子大小20-100nm;中强酸对应的NH3脱附峰温度为344℃,中强酸位点的量是2mol/kg。
作为对比例13,制备过程与实施例16基本一致,不同之处在于更换了多元金属复合物中的金属元素,保证替换金属的与被替换金属的原子数量相等,在配置金属前驱体溶液时将硝酸锌替换成硝酸铈。
作为对比例14,制备过程与实施例16基本一致,不同之处在于更换了多元金属复合物中的金属元素,保证替换金属的与被替换金属的原子数量相等,在配置金属前驱体溶液时将硝酸锌替换成纳米二氧化钛溶胶。
在400℃,3MPa体积空速4000h-1的反应条件下,反应结果如下表。
对比例13,14不含有权利要求的必要元素,所以几乎没有催化活性。
实施例17
多级孔结构的硅磷铝无机固体酸制备过程和方法与实施例5相同,不同之处在于金属前驱体溶液配置是分别称取六水合硝酸锌1.73g,按照元素摩尔比为Zn:Ga=1:1,称取硝酸镓1.49g,溶于100ml去离子水中,按照元素摩尔比Zn:Ti=1:1称取并加入TiO2 4.65g。
氮气物理吸脱附表征显示该催化剂具有多级孔结构,微孔比表面为289m2/g,微孔孔径大小分布在介孔比表面积为267m2/g,大孔比表面为161m2/g,介孔孔径大小分布在5-13nm。X射线衍射(XRD)表征显示,多组分金属复合物含ZnO、Ga2O3、TiO2呈CHA结构的硅铝无机固体酸,金属氧化物的晶粒大小13-20nm,无机固体酸晶粒大小为60-80nm;扫描电镜(SEM)表征表面,硅铝无机固体酸二次粒子大小为300-700μm;高分辨透射电镜(HRTEM)表征表明,多组分金属复合物的晶粒大小13-20nm,二次粒子大小30-120nm;中强酸对应的NH3脱附峰温度为336℃,中强酸位点的量是0.47mol/kg。
实施例18
制备过程和方法与实施例3基本相同,不同之处在于制取多级孔结构的硅磷铝无机固体酸,取六水合硝酸锌1.66g、按照元素摩尔比为Zn:Co:Mn:Ti=1:0.05:1:1称取六水合硝酸钴,0.5%wt硝酸锰水溶液,氯化钛配成混合液A,称取15.41g碳酸铵,溶解于100ml水中配成溶液B。
氮气物理吸脱附表征显示该催化剂具有多级孔结构,微孔比表面为316m2/g,微孔孔径分布在介孔比表面积为140m2/g,大孔比表面为99m2/g,介孔孔径大小分布在3-9nm。X射线衍射(XRD)表征显示,多组分金属复合物含Co2O3、Co3O4、MnO2、TiO2、ZnCo2O4、ZnMnO3和CHA结构的硅铝无机固体酸,金属氧化物晶粒大小9-18nm,无机固体酸晶粒大小为50-60nm;扫描电镜(SEM)表征表明,硅铝无机固体酸二次粒子大小为100-500μm;高分辨透射电镜(HRTEM)表征表明,多组分金属复合物的晶粒大小9-18nm,二次粒子大小20-100nm;中强酸对应的NH3脱附峰温度为368℃,中强酸位点的量是2.3mol/kg。
将实施例17、18的催化剂在400℃,3.5MPa体积空速4000h-1的反应条件下,实施例17、18反应结果如下表。
实施例19,将实施例17的催化剂在400℃,3.5MPa体积空速1500h-1的反应条件下,结果作为实施例19的反应结果列在下表。
实施例20,将实施例18的催化剂在400℃,3.5MPa体积空速1500h-1的反应条件下,结果作为实施例20的反应结果列在下表。
对比例15,将实施例17的催化剂在400℃,3.5MPa体积空速300h-1的反应条件下,结果作为对比例15的反应结果列在下表。
对比例16,将实施例18的催化剂在400℃,3.5MPa体积空速300h-1的反应条件下,结果作为对比例16的反应结果列在下表。
实施例17-20与对比例15-16,在不改变催化剂的前提下,只是改变了反应空速条件,其中实施例17-18的空速在权利要求的优选范围内,所以表现出了最高的低碳烯烃的选择性,而实施例19-20的空速虽然不在优选范围内,但属于权利要求的范围内所以仍有较高的低碳烯烃的选择性,而相对的对比例15-16,因为其反应空速不在权利要求范围内所以低碳烯烃选择性很低。
实施例21
化学复合法制备多组分金属复合物-多级孔结构的硅铝无机固体酸复合催化剂
以CaMnAl多组分金属复合物,多级孔结构的硅铝无机固体酸为例。
按氧化物SiO:Al2O3:Ca2O:R2O:H2O=40:1:7:5:900(质量比)称取原料:硅溶胶30%(质量浓度);硫酸铝;碳酸钙;N,N,N-一三甲基金刚烷氢氧化铵(R);去离子水,室温混合后,在60℃下,200rpm速度搅拌24h后转移到水热釜中,155℃下晶化6天。自然冷却到室温,转移到烧杯中水浴70℃,按100ml原液: 4g氯化铵的比例加入氯化铵,恒温搅拌3h,反复离心洗涤使得洗涤结束时上清液pH是7,沉淀物于120℃下烘干24h后,在650℃空气中焙烧3h得到多级孔结构的硅铝无机固体酸。
称取制备好的多级孔结构的硅铝无机固体酸,称取四水合硝酸锰1.49g,按照元素摩尔比为Mn:Ca:Al=1:0.1:1称取无水硝酸钙,九水合硝酸铝,溶解于100ml水溶液中,通过浸渍法将CaMnAl引入到多级孔结构的硅铝无机固体酸上,并经过室温真空干燥,520℃静态空气中焙烧1.5h,得到Ca0.1Mn1Al1/mesoSiAl。多组分金属复合物的负载量为50wt%。根据此方法,可以改变各金属组分及其比例,以及多组分金属复合物的负载量。
氮气物理吸脱附表征显示该催化剂具有多级孔结构,总比表面积565m2/g,其中微孔比表面占总比表面积的27%,微孔孔径分布在介孔比表面积占总比表面积57%,大孔比表面占总比表面积16%;介孔孔径大小分布在6-12nm。X射线衍射(XRD)表征显示,多组分金属复合物含CaO、MnO2、Al2O3、和具有CHA结构的硅铝无机固体酸,金属氧化物的晶粒大小5-13nm;无机固体酸晶粒大小为40-70nm;扫描电镜(SEM)表征表明,多级孔结构的硅铝无机固体酸二次粒子大小为10-100μm,无机固体酸晶粒大小为10-100nm;高分辨透射电镜(HRTEM)表征表明,多组分金属复合物的晶粒大小5-13nm,二次粒子大小15-50nm;中强酸对应的NH3脱附峰温度为370℃,中强酸位点的量是6mol/kg。
实施例22
催化剂制备过程与实施例21基本相同,不同之处在于制备多级孔结构的硅铝无机固体酸过程中,称取并室温混合前驱体后,控制搅拌时间为18h后转移到水热釜中,170℃晶化5天。
获得的多级孔催化剂,总比表面积493m2/g,其中微孔比表面占总比表面积的24%,微孔孔径分布在介孔比表面积占总比表面积53%,大孔比表面占总比表面积23%;介孔孔径大小分布在5-11nm。中强酸对应的NH3脱附峰温度为392℃,中强酸位点的量是3.1mol/kg。
实施例23
催化剂制备过程与实施例21基本相同,不同之处在于制备多级孔结构的硅铝无机固体酸过程中,称取并室温混合前驱体后,控制控制搅拌时间为3h后转移到水热釜中,165℃晶化5天又10h。最后在620℃焙烧80min。
获得的多级孔催化剂,总比表面积408m2/g,其中微孔比表面占总比表面积的16%,微孔孔径分布在介孔比表面积占总比表面积53%,大孔比表面占总比表面积31%;介孔孔径大小分布在5-13nm。中强酸对应的NH3脱附峰温度为389℃,中强酸位点的量是3.4mol/kg。
对比例17
催化剂制备过程与实施例21基本相同,不同之处在于制备多级孔结构的硅铝无机固体酸过程中,称取并室温混合前驱体后,控制控制搅拌温度90时间为1h后转移到水热釜中,215℃晶化7天。最后在550℃焙烧4h。
获得的多级孔催化剂,总比表面积390m2/g,其中微孔比表面占总比表面积的66%,微孔孔径分布在介孔比表面积占总比表面积26%,大孔比表面占总比表面积8%;介孔孔径大小分布在2-4nm。中强酸对应的NH3脱附峰温度为403℃,中强酸位点的量是4.1mol/kg。
对比例18
催化剂制备过程与实施例21基本相同,不同之处在于制备多级孔结构的硅铝无机固体酸过程中,称取并室温混合前驱体后,控制控制搅拌温度30时间为48h后转移到水热釜中,175℃晶化8天。最后在550℃焙烧3h。
获得的多级孔催化剂,总比表面积314m2/g,其中微孔比表面占总比表面积的81%,微孔孔径分布在介孔比表面积占总比表面积14%,大孔比表面占总比表面积5%;介孔孔径大小分布在2-4nm。中强酸对应的NH3脱附峰温度为368℃,中强酸位点的量是0.9mol/kg。
在375℃,3.7MPa体积空速6000h-1的反应条件下,反应结果如下表
实施例21-23与对比例17-18比较表现出了更优越的低碳烯烃的选择性,这是因为对比例17-18中三级孔道含量分布不符合权利要求的范围,而实施例22,23比实施例21有着更高的低碳烯烃的选择性,这是由于实施例21的大孔含量不在优选范围,且实施例22-23的三级孔含量不仅在优选范围更属于更优选的范围所以有着卓越的性能。
实施例24
化学复合法制备多组分金属复合物-多级孔结构的硅磷铝无机固体酸复合催化剂
按氧化物MgO:SiO2:Al2O3:H3PO4:R:H2O=2:18:16:32:55:150(质量比)称取原料:白炭黑;硝酸铝;磷酸;TEAOH(R);去离子水,室温混合后,在30℃下,500rpm速度搅拌10h后转移到水热釜中,200℃下晶化29h晶化中维持搅拌,搅拌速度100rpm。水浴骤冷到室温,反复离心洗涤使得洗涤结束时上清液pH是7,沉淀物于110℃下烘干24h后,在600℃空气中焙烧3h得到多级孔结构的硅磷铝无机固体酸。
使用上面方法制得的多级孔结构的硅磷铝无机固体酸,并称取四水合硝酸锰1.54g、按照元素摩尔比为Mn:Ca:Cr=2:0.2:1称取氯化钙、九水合硝酸铬,溶解于水溶液中配成混合液A,其中无机固体酸与Cr质量比2:1,称取6.3g碳酸铵,溶解于100ml水中配成溶液B,50℃下将溶液B逐滴加入以7W功率tip超声,400rpm速度搅拌混合液A中,离心洗涤使得洗涤结束时上清液pH是7并经过110℃空气中干燥,500℃静态空气中焙烧1h,得到Ca0.2Mn2Cr1/MgSiPAl。然后,以此为载体,准确称取醋酸钯Co(Ac)2溶解在水中,通过浸渍引入到CaMnCr/MgSiPAl上,其中Co与Mn的摩尔比为0.01。根据此方法,可以改变各金属组分及其比例,以及多元金属复合物的负载量。
氮气物理吸脱附表征显示该催化剂具有多级孔结构,微孔孔径分布在 介孔孔径大小分布在4-8nm,总的比表面积为416m2/g,其中微孔比表面积占57%,介孔比表面积占总比表面积的21%,大孔比表面占总面积22%,硅铝无机固体酸二次粒子大小为100-500μm,其晶粒大小为5-70nm;高分辨透射电镜(HRTEM)表征表明,多组分金属复合物的晶粒大小0.5-16nm,二次粒子大小10-100nm,;中强酸对应的NH3脱附峰温度为371℃,中强酸位点的量是2.1mol/kg。
实施例25
催化剂制备过程与实施例21基本相同,不同之处在于制备多级孔结构的硅铝无机固体酸过程中,称取并室温混合前驱体后,控制搅拌温度40℃,时间为2h后转移到水热釜中,180℃晶化2天。最后在流动空气下550℃焙烧4h。
氮气物理吸脱附表征显示该催化剂具有多级孔结构,微孔孔径分布在介孔孔径大小分布在5-9nm,总的比表面积为357m2/g,其中微孔比表面积占43%,介孔比表面积占总比表面积的30%,大孔比表面占总面积27%,硅铝无机固体酸二次粒子大小为50-150μm,其晶粒大小为20-80nm;高分辨透射电镜(HRTEM)表征表明,多组分金属复合物的晶粒大小1-13nm,二次粒子大小10-80nm;中强酸对应的NH3脱附峰温度为358℃,中强酸位点的量是1.9mol/kg。
实施例26
催化剂制备过程与实施例21基本相同,不同之处在于制备多级孔结构的硅铝无机固体酸过程中,称取并室温混合前驱体后,控制搅拌温度为50℃,时间为1h后转移到水热釜中,165℃晶化5天又10h。最后在静态空气620℃焙烧2h。
氮气物理吸脱附表征显示该催化剂具有多级孔结构,微孔孔径分布在介孔孔径大小分布在4-9nm,总的比表面积为4415m2/g,其中微孔比表面积占33%,介孔比表面积占总比表面积的35%,大孔比表面占总面积32%,硅铝无机固体酸二次粒子大小为50-100μm,其晶粒大小为40-90nm;高分辨透射电镜(HRTEM)表征表明,多组分金属复合物的晶粒大小3-6nm,二次粒子大小10-90nm;中强酸对应的NH3脱附峰温度为397℃,中强酸位点的量是2.7mol/kg。
对比例19
催化剂制备过程与实施例21基本相同,不同之处在于制备多级孔结构的硅铝无机固体酸过程中,按氧化物MgO:SiO2:Al2O3:H3PO4:R:H2O:2:18:16:32:10:150(质量比),称取原料称取并室温混合前驱体。控制晶化温度为150℃,时间24h。
氮气物理吸脱附表征显示该催化剂具有多级孔结构,微孔孔径分布在介孔孔径大小分布在4-9nm,总的比表面积为95m2/g,其中微孔比表面积占11%,介孔比表面积占总比表面积的55%,大孔比表面占总面积34%;中强酸对应的NH3脱附峰温度为320℃,中强酸位点的量是0.02mol/kg。
对比例20
催化剂制备过程与实施例21基本相同,不同之处在于制备多级孔结构的硅铝无机固体酸过程中,按氧化物MgO:SiO2:Al2O3:H3PO4:R:H2O:2:18:16:32:80:150(质量比),称取原料称取并室温混合前驱体。控制晶化温度为180℃,时间72h。控制焙烧温度550℃,静态空气中,时间30min。
氮气物理吸脱附表征显示该催化剂具有多级孔结构,微孔孔径分布在 介孔孔径大小分布在4-9nm,总的比表面积为69m2/g,其中微孔比表面积占14%,介孔比表面积占总比表面积的24%,大孔比表面占总面积62%;中强酸对应的NH3脱附峰温度为329℃,中强酸位点的量是0.03mol/kg。
在415℃,3.0MPa体积空速5000h-1的反应条件下,反应结果如下表
实施例24-26所述的催化剂,多组分金属复合物及硅铝无机固体酸的晶粒及二次粒子的大小满足权利要求所述的范围,其比表面积满足权利要求的范围,并且形成了权利要求所述的三维三级孔道且三级孔道含量符合权利要求所述优选范围,其中实施例25-26在更优选范围,所以均表现出优异的性能。而对比例19-20虽然形成了三维三级孔道,但因为有机模板剂与水的比例不满足权利要求的范围,对比例19有机模板剂过少不利于形成足够的孔道结构,而对比例20有机模板剂过量,且焙烧时间不满足权利要求所述的时间范围,均导致了对比例19-20的比表面积低于权利要求的范围,从而表现为转化率很低,同时低碳烯烃的选择性也远低于本发明的催化剂。
实施例27
催化剂制备过程与实施例5基本相同,不同之处在于更换了多元金属复合物中的金属元素,在配置金属前驱体溶液时将六水合硝酸铜用碳酸氢钠进行替换,保证替换金属的原子与被替换金属的原子为(1:10),六水合硝酸钴用柠檬酸铁铵替换,保证替换金属的原子与被替换金属的原子为(1:2)。
实施例28
催化剂制备过程与实施例27基本相同,不同之处在于更换了多元金属复合物中的金属元素,在配置金属前驱体溶液时将碳酸氢钠用碱式碳酸镁进行替换,保证替换金属的原子与被替换金属的原子为(1:1)。
实施例29
催化剂制备过程与实施例27基本相同,不同之处在于更换了多元金属复合物中的金属元素,在配置金属前驱体溶液时将碳酸氢钠用硝酸钾进行替换,保证替换金属的原子与被替换金属的原子为(1:1)。
实施例30
催化剂制备过程与实施例27基本相同,不同之处在于更换了多元金属复合物中的金属元素,在配置金属前驱体溶液时将碳酸氢钠用硝酸铯进行替换,保证替换金属的原子与被替换金属的原子为(1:1)。
实施例31
催化剂制备过程与实施例27基本相同,不同之处在于更换了多元金属复合物中的金属元素,在配置金属前驱体溶液时将碳酸氢钠用硝酸镧进行替换,保证替换金属的原子与被替换金属的原子为(1:1)。
实施例32
催化剂制备过程与实施例5基本相同,不同之处在于更换了多元金属复合物中的金属元素,在配置金属前驱体溶液时将六水合硝酸铜用25wt.%硝酸锰进行替换,保证替换金属的与被替换金属的原子为(1:1),六水合硝酸钴用柠檬酸铁铵替换,保证替换金属的原子与被替换金属的原子为(1:2),γ-Al2O3用硝酸镓替换保证替换金属的原子与被替换金属的原子为(1:1)。
实施例33
催化剂制备过程与实施例32基本相同,不同之处在于更换了多元金属复合物中的金属元素,在配置金属前驱体溶液时将硝酸镓用硝酸锗进行替换,保证替换金属的原子与被替换金属的原子为(1:1)。
实施例34
催化剂制备过程与实施例32基本相同,不同之处在于更换了多元金属复合物中的金属元素,在配置金属前驱体溶液时将硝酸镓用硝酸锆进行替换,保证替换金属的原子与被替换金属的原子为(1:1)。
实施例35
催化剂制备过程与实施例32基本相同,不同之处在于更换了多元金属复合物中的金属元素,在配置金属前驱体溶液时将硝酸镓用硝酸铟进行替换,保证替换金属的原子与被替换金属的原子为(1:1)。
实施例36-40
催化剂制备过程与实施例27-31基本相同,不同之处在于更换了多元金属复合物中的金属元素,在配置金属前驱体溶液时将硝酸锌用25wt.%硝酸锰进行替换,保证替换金属的原子与被替换金属的原子为(1:1)。
在400℃,3.0MPa体积空速6000h-1的反应条件下,反应结果如下表。
对比例21
作为对比例21,商品Na-ZSM-5分子筛与共沉淀法制备的Co0.1Cr3Zn2的复合氧化物机械混合得到的混合物作为催化剂,在相同条件下,低碳烷烃及烯烃的选择性不超过20%,产生大量甲烷而且催化剂的稳定性极差,15小时随即失活。这是由于该分子筛仅含微孔结构,不具有介孔结构,反应生成的积碳极易堵塞微孔,从而导致失活。
对比例22
作为对比例22,用于甲醇合成的商品CuZnAl催化剂与不具备多级孔结构的商品MTO催化剂SAPO-34,按照质量比1:1,进行机械混合,在相同条件下反应。只能得到烷烃,几乎没有烯烃生成。这是因为商品SAPO-34分子筛不具备权利要求所述的多级孔结构,不利于传质,导致产物过度加氢,使得产物中几乎没有烯烃产生。
对比例23
以碳纳米管为载体,管腔内负载FeN纳米颗粒,可以限制纳米粒子的聚集长大,避免失活,经过K和Mn促进,得到K0.4Mn1.0FeN@CNTs,可以选择性生成低碳烯烃。在300℃,5bar,空速15000h-1,H2/CO/Ar=47.5/47.4/5.1的条件下,转化率为11.9%,C2=-C4=的选择性为43.6%。在该催化剂表面,CO解离生成的表面碳物种,与解离的氢原子反应生成CHx中间物种,在表面上进一步相互偶联,生成不同碳链长度的碳氢化合物或者加氢生成烷烃,因此产物众多,产物符合ASF分布。
在400℃,2MPa体积空速4000h-1的反应条件下,对比例21、22反应结果如下表。
在300℃,0.5MPa体积空速15000h-1的反应条件下,对比例23反应结果如下表。
实施例1-40所述催化剂的反应产物中产物中C2-C4在总烃产物中的选择性可达60~95%,低碳烯烃在总烃产物中的选择性高达50~85%,由于催化剂金属复合物表面不活化氢分子,避免了甲烷的大量生成,甲烷选择性和C5+长碳链碳氢化合物的选择性可以极低,两者小于10%。无机固体酸的多级孔结构避免催化剂失活,连续反应100小时CO的转化率稳定,产物选择性稳定。
实施例1-40中催化剂的性能明显优于传统改进的费托催化剂,以及传统MTO反应催化剂与甲醇合成催化剂机械混合得到的催化剂。原因是由于本发明的催化剂金属复合物表面经过含氢气或一氧化碳的气氛还原活化之后,在表面结构中形成氧空位,即氧为配位不饱和状态,可高效地催化活化CO生成中间态物种CHx(其中x=1,2,3)中的一种或两种以上的物种,表面氧物种与CO反应生成CO2,这些极其活泼的CHx物种与CO结合生成CHxCO,避免了甲烷的大量生成,甲烷选择性和C5+长碳链碳氢化合物的选择性极低,两者共为5~10%。同时生成的中间态物种在表面为弱吸附,极易从表面脱附并扩散至无机固体酸的孔道内,被酸性位进一步催化转化反应生成低碳烃。本发明的无机固体酸具有多级孔结构,从而避免催化剂失活,连续反应100小时CO的转化率稳定,产物选择性稳定。而传统微孔MTO分子筛催化剂不具备多级孔结构,很容易被反应生成的中间物种堵塞导致催化剂快速失活,且不利于传质直接混合使用只能得到烷烃,几乎没有烯烃产生。相比之下,Fe为主成分的催化剂仍遵循费托反应机理,即CO在催化剂表面解离吸附,生成表面碳和表面氧,表面氧与吸附态的氢反应生成水,表面碳则与表面氢反应生成CHx,CHx在表面吸附较强,多个表面CHx聚合,发生链增长,由催化剂表面性质决定形成不同碳链长度的碳氢化合物并脱附至气相。
本发明所提供的催化剂为复合材料,其有效成分包括多组分金属复合物和多级孔结构的无机固体酸,可以催化合成气转化生成C2-C4低碳烃,CO单程转化率为10~60%,低碳烃在所有碳氢化合物产品中的选择性可高达60~95%,其中低碳烯烃(C2=-C4=)的选择性为50~85%。该方法具有工艺简单,设备投资少的特点,具有重要的应用前景。
- 还没有人留言评论。精彩留言会获得点赞!