氢化硅烷化反应催化剂的制作方法
本发明涉及氢化硅烷化催化剂,进一步详细地说,涉及由作为催化剂前体的金属化合物和作为配体成分的卡宾化合物形成的氢化硅烷化催化剂。
背景技术:
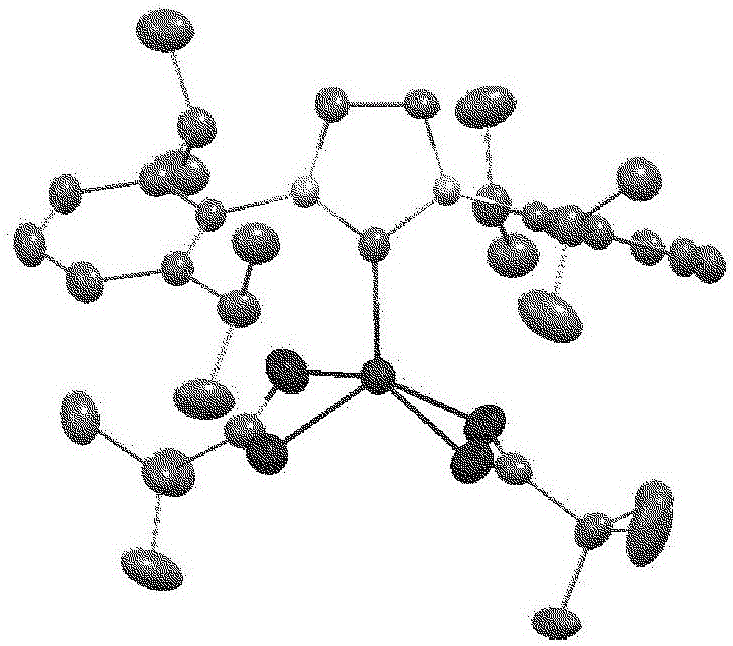
:使Si-H官能性化合物对于具有碳-碳双键、碳-碳三键的化合物进行加成反应的氢化硅烷化反应是合成有机硅化合物的有用的手段,在工业上也是重要的合成反应。作为该氢化硅烷化反应的催化剂,已知Pt、Pd、Rh化合物,其中最多地使用的是以Speier催化剂、Karstedt催化剂为代表的Pt化合物。作为以Pt化合物作为催化剂的反应的问题,可列举出使Si-H官能性化合物加成于末端烯烃时发生烯烃内部重排的副反应。在该体系中对于内部烯烃不显示加成反应性,未反应烯烃在加成生成物中残留,因此为了使反应完结,必须通过预估由于副反应而残留的部分来使用过量的烯烃。另外,也存在根据烯烃的种类、α加成体和β加成体的选择性差的问题。最大的问题在于作为中心金属的Pt、Pd、Rh都是价格极高的贵金属元素,从希望有能够更低价地使用的金属化合物催化剂的角度,进行了大量的研究。例如,已知采用铁-羰基络合物(Fe(CO)5、Fe3(CO)12)的反应(非专利文献1),该反应中如160℃这样的高温下的反应条件、或光照射(非专利文献2)是必要的。另外,对于这些铁-羰基络合物,也报道了不是加成反应而是得到脱氢硅烷化生成物(非专利文献3、专利文献1)。也报道了使用了具有环戊二烯基作为配体的铁-羰基络合物的、甲基乙烯基二硅氧烷和甲基氢二硅氧烷的反应实例(非专利文献4、专利文献2),但在该反应中由于脱氢硅烷化反应也进行,因此加成反应的选择性低。在具有三联吡啶系配体的铁催化剂的反应中(非专利文献5),不仅作为反应助剂需要大幅过量的还原剂(NaBHEt3),而且虽然PhSiH3、Ph2SiH2对于烯烃进行加成,但有用性更高的三烷基硅烷类、烷氧基硅烷类、硅氧烷类对于烯烃的加成反应性缺乏。虽然报道了同样地通过具有三联吡啶系配体和双(三甲基甲硅烷基)甲基的铁催化剂的反应收率良好地得到加成反应物(非专利文献6),但该方法首先合成成为催化剂前体的三联吡啶-铁络合物,进而低温下的双(三甲基甲硅烷基)甲基的导入和直至催化剂合成在工业上并不容易。也报道了具有双亚氨基吡啶配体的Fe络合物(非专利文献7、8),公开了即使对于烷氧基硅烷类、硅氧烷类在温和的条件下也显示优异的反应性。但是,在使用了该络合物的反应中,具有如下问题:对于内部烯烃的反应性低,络合物合成时使用由禁水性的钠和毒性高的水银构成、处理上需要注意的Na汞齐(或者使用禁水性的NaBEt3H),配位化合物自身的稳定性低,处理需要手套箱等特殊的设备,此外保存必须为非活性气体氮气氛下并且为低温。也报道了采用钴-羰基络合物(Co2(CO)8等)的反应实例(非专利文献9~14),但在反应收率、反应摩尔比的方面并未满足要求,对于与硅氧烷类的加成反应性也没有公开。也报道了采用具有三烷基甲硅烷基作为取代基的钴-羰基络合物的烯烃与三烷基硅烷的反应实例(非专利文献15),但收率低,另外也缺乏选择性。报道了通过使用具有环戊二烯基作为配体的钴-亚磷酸酯络合物而进行的烯烃与三烷基硅烷的反应(非专利文献16)、采用具有N-杂环状卡宾作为配体的钴络合物的烯烃与三氢苯基硅烷的反应(非专利文献17),但配位化合物的稳定性低,处理需要手套箱等特殊的设备,保存需要在非活性气体气氛下并且低温下。也报道了使配体为三联吡啶、双亚氨基吡啶、双亚氨基喹啉的、铁、钴、镍催化剂的例子(专利文献3~6),但与上述的非专利文献6~8同样地存在如下的问题:催化剂前体的合成、或者从该前体直至络合物催化剂的合成在工业上并不容易,配位化合物自身的稳定性低,处理需要特殊的设备。另外,也公开了在采用具有双亚氨基喹啉配体的络合物催化剂的反应中使用Mg(丁二烯)·2THF、NaEt3BH作为催化剂的活化剂的方法(专利文献7),除了存在与上述相同的问题以外,目标物的收率也未能满足要求。也报道了大量的镍络合物催化剂。例如,对于以膦作为配体的催化剂(非专利文献18),选择性差,催化剂的保存、处理需要注意。就乙烯基硅氧烷配位的催化剂(非专利文献19)而言,脱氢硅烷化生成物成为主成分,加成反应的选择性低。就以烯丙基膦作为配体的催化剂(非专利文献20)而言,收率低,三氢苯基硅烷并不是工业上价值高的反应基质。就具有双酰氨基的催化剂(非专利文献21)而言,必须注意催化剂的保存、处理,另外二氢二苯基硅烷也不是工业上价值高的反应基质。就具有N-杂环状卡宾作为配体的催化剂(非专利文献22)而言,反应的选择性低,三氢苯基硅烷的工业上的价值不高。也报道了大量的铑络合物催化剂。例如,就具有羰基或COD基(环辛二烯基)和N-杂卡宾配体的催化剂(非专利文献23、24)而言,都是配位化合物的稳定性低,因此必须在非活性气体气氛下处理和保存。也公开了为了提高反应性而在离子性液体存在下反应的方法(非专利文献25),但需要从反应生成物中将离子性液体分离的工序。另外,催化剂也具有COD基和N-杂卡宾基作为配体,具有与上述相同的问题。另外,也报道了优先进行脱氢硅烷化反应的催化剂的例子(非专利文献26)。进而,也报道了在络合物催化剂中加入异腈化合物、在没有将生成的催化剂离析的情况下进行了氢化硅烷化的例子(非专利文献27)。对于与3种硅烷的反应性进行研究,就反应性而言,二甲基苯基硅烷为最高收率(收率81%),其次为三乙基硅烷(收率66%)、三乙氧基硅烷(收率40%),这3种硅烷中工业上价值最高的三乙氧基硅烷中的反应性不高,另外,对于与硅氧烷的反应性没有报道。进而,成为前体的催化剂以COD基作为配体,对于保存、处理需要注意。另一方面,也报道了用具有乙酰丙酮基或乙酸酯基的铑催化剂、三乙氧基硅烷收率良好地进行加成反应(非专利文献28)。该方法虽然具有催化剂的保存和处理容易的优点,但对于与从工业的观点出发实用性更高的硅氧烷的反应性没有研究。并且,不变的是,铑也为高价的贵金属元素,要作为替代铂的催化剂实用化要求进一步使催化剂功能高活性化。作为关注向有机聚硅氧烷的应用的实例,公开了具有膦配体的催化剂(专利文献8)、具有芳基-烷基-三氮烯基的催化剂(专利文献9)、胶体状的催化剂(专利文献10)、以硫醚基作为配体的催化剂(专利文献11)、以氨基、膦基、硫醚基和有机硅氧烷基作为配体的催化剂(专利文献12)。但是,具体地例示了反应活性的仅为作为高价的金属元素的铂、钯、铑、铱,不能称得上是成本上有利的方法。另外,在专利文献13、14的实施例中示出了效果的仅为公知的铂催化剂,对于用其他的金属显示催化剂活性的结构没有任何教导。也公开了具有卡宾作为配体的催化剂(专利文献15~17),但在专利文献15中对于对氢化硅烷化反应的有效性没有任何研究。专利文献16和专利文献17中公开了具有卡宾和乙烯基硅氧烷作为配体的催化剂,但作为实施例记载的只是铂催化剂。并且,就具有卡宾作为配体的金属催化剂而言,配位化合物的保存稳定性低,处理也需要注意。同样地作为具有卡宾作为配体的催化剂的例子,专利文献27、28中只公开了铂催化剂。另外,专利文献29中公开了由Ni-卡宾络合物与金属前体的反应得到的金属-卡宾络合物催化剂。但是,必须另外合成Ni-卡宾络合物,反应的金属前体是具有膦、COD等配体的金属化合物,具有这些配体的金属前体的保存稳定性低。专利文献30、31中公开了使具有烯烃性的配体的Pd、Pt、Ni络合物与卡宾反应而得到的络合物催化剂。但是,具有烯烃性配体的金属络合物除了以公知的乙烯基硅氧烷作为配体的Pt催化剂以外,保存稳定性低。专利文献32中公开了Co-卡宾络合物,其在对于酮类的氢化硅烷化反应中具有活性。专利文献33、34中公开了在采用金属-卡宾络合物的有机聚硅氧烷的固化反应中的应用,但其中所示的金属只是Pt,合成方法也是公知的具有乙烯基硅氧烷作为配体的Pt络合物与卡宾反应。也公开了具有η6-芳烃、η6-三烯作为配体的钌催化剂(专利文献18、19),但与铂催化剂相比,反应活性差,另外配位化合物的保存稳定性低,处理也需要注意。也公开了如下方法:不是以金属络合物作为催化剂,而是混合金属盐和对于金属具有配位性的化合物,将其用作催化剂(专利文献20~26)。但是,这些专利文献中,虽然记载了以几例的组合进行了氢化硅烷化,但无收率的记载等,反应有效地进行到何种程度不清楚。例如,虽然专利文献21、22中分别记载了几个在Co、或Fe的卤化物、或三甲基甲硅烷基酰胺盐中添加了相当于卡宾的化合物的实施例,但记载为具有反应活性的只是苯基三氢硅烷,对于七甲基三硅氧烷,记载了无反应活性。同样地在专利文献25中公开了Ni化合物和卡宾化合物的例子,但在与七甲基三硅氧烷的加成反应中确认了活性的只是1例,此外示出了大量只对苯基三氢硅烷显示出活性、或者对七甲基三硅氧烷也不显示活性的例子。专利文献23、26中公开了Ir、或Ru化合物和卡宾化合物的例子,但显示反应活性的只是具有COD基、或η6-芳基作为烯烃性的配体的金属化合物。并且,这些专利文献21~26中所示的全部的实施例都使用了离子性的盐或氢化物还原剂作为活化剂,尽管如此,在几乎全部实施例中都没有显示催化剂活性。现有技术文献专利文献专利文献1:国际公开第2013/081794号专利文献2:国际公开第2010/016416号专利文献3:日本特表2012-532885号公报专利文献4:日本特表2012-532884号公报专利文献5:日本特表2013-544824号公报专利文献6:日本特表2014-502271号公报专利文献7:日本特表2014-503507号公报专利文献8:日本特开平6-136126号公报专利文献9:日本特开平6-263780号公报专利文献10:日本特开平1-315344号公报专利文献11:日本专利第3174616号公报专利文献12:日本特开平7-149780号公报专利文献13:日本特开2001-131231号公报专利文献14:日本专利第4007467号公报专利文献15:日本专利第3599669号公报专利文献16:日本专利第3854151号公报专利文献17:日本专利第4249702号公报专利文献18:日本专利第4934190号公报专利文献19:日本专利第5032561号公报专利文献20:国际公开第2013/043846号专利文献21:国际公开第2013/043783号专利文献22:国际公开第2013/043912号专利文献23:国际公开第2014/021908号专利文献24:国际公开第2013/081794号专利文献25:国际公开第2013/043785号专利文献26:国际公开第2013/043787号专利文献27:中国专利申请公开第102516314号说明书专利文献28:美国专利申请公开第2011/0160454号说明书专利文献29:中国专利申请公开第102351907号说明书专利文献30:国际公开第2008/095785号专利文献31:法国专利发明第2911876号说明书专利文献32:美国专利第6737531号说明书专利文献33:美国专利申请公开第2004/0236054号说明书专利文献34:美国专利第7019145号说明书非专利文献非专利文献1:A.N.Nesmeyanov等,Tetrahedron,1962,17,61非专利文献2:M.S.Wrighton等,J.Organomet.Chem.,1977,128,345非专利文献3:F.Kakiuchi等,J.Organomet.,Chem.,1993,456,45非专利文献4:H.Nakazawa等,J.Am.Chem.Soc.,2012,134,804非专利文献5:H.Nakazawa等,Organometallics,2012,31,3825非专利文献6:P.J.Chirik等,Organometallics,2012,31,4886非专利文献7:P.J.Chirik等,J.Am.Chem.Soc.,2004,126,13794非专利文献8:P.J.Chirik等,Science,2012,335,567非专利文献9:A.J.Chalk等,J.Am.Chem.Soc.,1965,87,1133非专利文献10:A.J.Chalk等,J.Am.Chem.Soc.,1967,89,1640非专利文献11:A.J.Chalk,J.Organomet.Chem.,1970,21,207非专利文献12:B.A.Izmailov等,J.Organomet.Chem.,1978,149,29非专利文献13:N.Sonoda等,J.Org.Chem.,1987,52,4864非专利文献14:S.Murai等,Chem.Lett.,2000,14非专利文献15:M.S.Wrighton等,Inorg.Chem.,1980,19,3858非专利文献16:B.E.Grant等,J.Am.Chem.Soc.,1993,115,2151非专利文献17:L.Deng等,Angew.Chem.Int.Ed.,2013,52,10845非专利文献18:M.Umeno等,J.Organomet.Chem.,1973,50,297非专利文献19:I.Kownacki等,J.Organomet.Chem.,2000,597,175非专利文献20:P.Valerga等,DaltonTrans.,2007,3000非专利文献21:T.D.Tilley等,Chem.Commun.,2012,48,7146非专利文献22:P.Valerga等,Organometallics,2012,31,2175非专利文献23:T.A.Nile等,J.Organomet.Chem.,1977,137,293非专利文献24:M.R.Buchmeiser等,J.Organomet.Chem.,2005,690,4433非专利文献25:X.Li等,J.Organomet.Chem.,2011,696,2116非专利文献26:S.P.Nolan等,DaltonTrans.,2013,42,270非专利文献27:J.M.Walters等,J.MolecularCatalysis,1985,29,201非专利文献28:M.F.Lappert等,J.Organomet.Chem.,1979,172,153技术实现要素:发明要解决的课题本发明鉴于这样的实际情况而完成,目的在于提供可在温和的条件下使氢化硅烷化反应进行、处理性和保存性优异的氢化硅烷化反应催化剂和使用了其的采用氢化硅烷化反应的加成化合物的制造方法。用于解决课题的手段本发明人为了实现上述目的进行了锐意研究,结果发现:使用指定的金属化合物作为催化剂前体、同时使用卡宾化合物作为配体成分得到了的催化剂对氢化硅烷化反应可发挥优异的活性,在温和的条件下进行加成反应,完成了本发明。即,本发明提供:1.氢化硅烷化反应催化剂,其特征在于,自由式(1)所示的金属盐化合物和由式(2)所示的、1个或2个氮原子邻接的卡宾化合物而制备:Ma(L)b(X)c(1){式(1)中,M表示除铂以外的周期表第8族、第9族、第10族的过渡金属,X表示卤素原子,L表示选自下述式(3)~(5)中的至少1种的1价有机基团,a表示1或2的整数,b表示0~6的整数,c表示0~3的整数,a为1时,b+c满足为2或3,a为2时,b+c满足4~6。-O-R1(3)-OCO-R1(4)-OSO2-R1(5)[式(3)~(5)中,R1相互独立地表示可被取代、并且、1个或1个以上的选自氧、氮、硫和磷中的原子可介于其间的碳原子数1~30的1价有机基团或由式(6)表示的1价的有机基团。-(A)p-R2(6)〈式(6)中,A表示可被卤素原子取代的碳原子数1~30的2价有机基团,p表示0或1的整数,上述L为由式(3)表示的1价有机基团时,p满足为0或1,上述L为由式(4)或式(5)表示的1价有机基团时,p满足为1,R2表示由式(7)表示的基团。-{Si(R3)2-R4}s-Si(R3)d{[(OSi(R3)2)]f-R3}e(7)(式(7)中,R3相互独立地表示可被取代、并且、1个或1个以上的选自氧、氮、硫和磷中的原子可介于其间的、碳原子数1~20的烷基、碳原子数1~20的烷氧基、碳原子数6~20的芳基、或碳原子数7~20的芳烷基,R4表示碳原子数1~10的2价烃基,s表示0或1的整数,d表示0~3的整数,e表示0~3的整数,并且d+e满足为3,f表示1~300的整数。)〉]}[化1](式(2)中,Y表示碳原子、氮原子或氧原子,Y为碳原子时,g为3,Y为氮原子时,g为2,Y为氧原子时,g为1,R4和R5相互独立地表示可被卤素原子或烷氧基取代的、碳原子数1~30的烷基、芳基或芳烷基,R4的任一个与R5的任一个可结合构成2价的有机基团而形成环状结构,形成环状结构时,可包含氮原子和/或不饱和键。);2.1的氢化硅烷化反应催化剂,其中,上述式(7)的s为0;3.1或2的氢化硅烷化反应催化剂,其中,上述a为1或2,b为2~4,c为0~1,a为1时,b+c满足为2,a为2时,b+c满足4或5;4.1~3的任一项的氢化硅烷化反应催化剂,其是在使具有脂肪族不饱和键的化合物与具有Si-H基的氢硅烷化合物或有机氢聚硅氧烷化合物进行氢化硅烷化反应的体系内制备的;5.1~4的任一项的氢化硅烷化反应催化剂,其中,上述式(2)所示的卡宾化合物由下述式(8)表示:[化2](式中,Z表示可含有氮原子和/或不饱和键的碳原子数2~5的2价的有机基团,R4和R5相互独立地表示可被卤素原子或烷氧基取代的、碳原子数1~30的烷基、芳基或芳烷基。)6.1~5的任一项的氢化硅烷化反应催化剂,其中,上述M为Fe、Co或Ni,a为1,b为2,c为0;7.1~5的任一项的氢化硅烷化反应催化剂,其中,上述M为Rh,a为2,b为4,c为0;8.1~5的任一项的氢化硅烷化反应催化剂,其中,上述M为Ru,a为2,b为4,c为1;9.1~8的任一项的氢化硅烷化反应催化剂,其中,上述L为由上述式(4)表示的1价的有机基团;10.加成化合物的制造方法,其特征在于,在1~9的任一项的氢化硅烷化反应催化剂的存在下,使具有脂肪族不饱和键的化合物与具有Si-H键的氢硅烷化合物或有机氢聚硅氧烷化合物进行氢化硅烷化反应;11.10的加成化合物的制造方法,其中,上述具有脂肪族不饱和键的化合物为具有烯基的有机聚硅氧烷。发明的效果成为本发明的氢化硅烷化反应催化剂的原料的金属化合物由于为市售品、或采用公知的方法合成,因此能够容易地得到。另外,本发明的催化剂具有如下优点:由于不具有羰基、η4-二烯基、η5-环戊二烯基、η6-芳烃、或三烯基作为配体,因此不必在低温下或非活性气体气氛中保存,或者不必在手套箱中进行计量等处理,是极易处理的催化剂,即使在空气中长时间暴露,反应活性也高。另一方面,作为配体成分的卡宾化合物也可以在室温下保存,处理时不需要使用特别的装置。为了使用金属化合物使反应活性种产生,通常必须加入在体系内将高原子价金属种还原的还原剂,但本发明中由于利用作为反应物的氢硅烷自身作为还原剂,因此不必加入还原剂,进行作为目标的采用氢化硅烷化的加成反应。由这些金属化合物和卡宾化合物制备的催化剂可以作为金属配位化合物在离析后使用,也可以在进行氢化硅烷化反应的体系内制备而不离析地使用。如果使用由这些金属化合物和卡宾化合物制备的催化剂,进行含有脂肪族不饱和基团的化合物与具有Si-H基的硅烷、或者聚硅氧烷的氢化硅烷化反应,则可以在室温~100℃以下的条件下进行加成反应。特别是与工业上有用的聚硅氧烷、和三烷氧基硅烷、二烷氧基硅烷的加成反应也良好地进行。应予说明,在公知文献中示出了在同一反应中与不饱和基团的加成反应、和采用脱氢硅烷化反应的含有不饱和基团的化合物生成的反应经常同时进行,但如果使用本发明的催化剂,则与不饱和基团的加成反应选择性地进行。并且,在对于以往的催化剂而言困难的与内部烯烃的反应中,可以得到与不饱和基团的向末端的移动相伴的加成反应物,在有机硅工业中有用性极高。附图说明图1为表示合成例5中得到的钴络合物A的X射线结晶结构解析结果的图。图2为合成例5中得到的钴络合物A的1H-NMR波谱图。图3为表示合成例6中得到的铁络合物B的X射线结晶结构解析结果的图。图4为合成例6中得到的铁络合物B的1H-NMR波谱图。图5为合成例7中得到的钴羧酸盐C的FT-IR光谱图。图6为合成例8中得到的钴羧酸盐D的FT-IR光谱图。图7为合成例9中得到的钴羧酸盐E的FT-IR光谱图。具体实施方式以下对本发明更为详细地说明。本发明涉及的氢化硅烷化反应催化剂由作为催化剂前体的式(1)所示的金属化合物、和作为配体的式(2)所示的卡宾化合物制备。Ma(L)b(X)c(1)[化3]式(1)中,M表示除铂以外的周期表第8族、第9族、第10族的过渡金属,其中,优选Fe、Co、Ni、Ru、Rh、Pd、Os、Ir,如果考虑金属盐的获得容易性、成本、和催化剂活性等,更优选Fe、Co、Ni、Ru、Rh、Os、Ir,进一步优选Fe、Co、Ru、Ni、Rh。X表示卤素原子,作为其具体例,可列举出氟、氯、溴、碘原子,优选氯原子、溴原子,更优选氯原子。L表示与过渡金属M用氧原子结合的1价有机基团,具体地,表示选自式(3)~(5)中的至少1种,优选由式(4)表示的1价有机基团。-O-R1(3)-OCO-R1(4)-OSO2-R1(5)式(3)~(5)中,R1相互独立地表示可被取代、并且、1个或1个以上的选自氧、氮、硫和磷中的原子可介于其间的碳原子数1~30的1价有机基团、或由式(6)表示的1价的有机基团。-(A)p-R2(6)作为碳原子数1~30的1价的有机基团,并无特别限定,优选碳原子数1~30的1价烃基。作为1价烃基,可列举出烷基、烯基、炔基、芳基、芳烷基等。作为烷基,可以是直链、分支链、环状的任一种,优选为碳原子数1~20的烷基,更优选为碳原子数1~10的烷基,作为其具体例,可列举出甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、正己基、正庚基、正辛基、正壬基、正癸基、正十一烷基、正十二烷基、正十三烷基、正十四烷基、正十五烷基、正十六烷基、正十七烷基、正十八烷基、正十九烷基、正二十烷基等直链或分支链烷基;环丙基、环丁基、环戊基、环己基、环庚基、环辛基、环壬基、降冰片基、金刚烷基等环烷基等。作为烯基,优选碳原子数2~20的烯基,作为其具体例,可列举出乙烯基、正-1-丙烯基、正-2-丙烯基、1-甲基乙烯基、正-1-丁烯基、正-2-丁烯基、正-3-丁烯基、2-甲基-1-丙烯基、2-甲基-2-丙烯基、1-乙基乙烯基、1-甲基-1-丙烯基、1-甲基-2-丙烯基、正-1-戊烯基、正-1-癸烯基、正-1-二十碳烯基等。作为炔基,优选碳原子数2~20的炔基,作为其具体例,可列举出乙炔基、正-1-丙炔基、正-2-丙炔基、正-1-丁炔基、正-2-丁炔基、正-3-丁炔基、1-甲基-2-丙炔基、正-1-戊炔基、正-2-戊炔基、正-3-戊炔基、正-4-戊炔基、1-甲基-正-丁炔基、2-甲基-正-丁炔基、3-甲基-正-丁炔基、1,1-二甲基-正-丙炔基、正-1-己炔基、正-1-癸炔基、正-1-十五碳炔基、正-1-二十碳炔基等。作为芳基,优选为碳原子数6~30的芳基,更优选为碳原子数6~20的芳基,作为其具体例,可列举出苯基、1-萘基、2-萘基、蒽基、菲基、邻-联苯基、间-联苯基、对-联苯基等。作为芳烷基,优选为碳原子数7~30的芳烷基,更优选为碳原子数7~20的芳烷基,作为其具体例,可列举出苄基、苯基乙基、苯基丙基、萘基甲基、萘基乙基、萘基丙基等。另外,这些基团在不损害本发明的氢化硅烷化反应催化剂的活性的范围内,1个或1个以上的选自氧、氮、硫和磷中的原子可介于其间。另外,碳原子数1~30的1价的有机基团可具有取代基,可在任意的位置具有相同或不同的多个取代基。作为上述取代基的具体例,可列举出氟原子、氯原子等上述的各种卤素原子、甲氧基、乙氧基、丙氧基等烷氧基、二烷基氨基等氨基等。式(6)中,A表示可被取代、并且、1个或1个以上的选自氧、氮、硫和磷中的原子可介于其间的碳原子数1~30的2价有机基团,p表示0或1的整数,上述L为由式(3)表示的1价有机基团时,p满足为0或1,上述L为由式(4)或式(5)表示的1价有机基团时,p满足为1。作为碳原子数1~30的2价有机基团,并无特别限定,但优选碳原子数1~30的2价烃基。作为2价烃基,可列举出亚烷基、亚芳基、亚芳烷基等。作为亚烷基,优选直链、分支链、环状的任一种,优选为碳原子数1~20的亚烷基,更优选为碳原子数1~10的亚烷基,作为其具体例,可列举出亚甲基、亚乙基、亚丙基、三亚甲基、亚正丁基、亚异丁基、亚仲丁基、亚正辛基、2-乙基亚己基、亚正癸基、亚正十一烷基、亚正十二烷基、亚正十三烷基、亚正十四烷基、亚正十五烷基、亚正十六烷基、亚正十七烷基、亚正十八烷基、亚正十九烷基、亚正二十烷基等直链或分支链亚烷基;1,4-亚环己基等亚环烷基等。作为亚芳基,优选为碳原子数6~30的亚芳基,更优选为碳原子数6~20的亚芳基,作为其具体例,可列举出邻-亚苯基、间-亚苯基、对-亚苯基、1,2-亚萘基、1,8-亚萘基、2,3-亚萘基、4,4′-亚联苯基等。作为亚芳烷基,优选为碳原子数7~30的亚芳烷基,更优选为碳原子数7~20的亚芳烷基,作为其具体例,可列举出-(CH2)k-Ar-(Ar表示碳原子数6~20的亚芳基,k表示1~10的整数。)、-Ar-(CH2)k-(Ar和k表示与上述相同的含义。)、-(CH2)k-Ar-(CH2)k-(Ar表示与上述相同的含义,k相互独立地表示与上述相同的含义。)等。R2表示由式(7)表示的甲硅烷基或聚有机硅氧烷基。-{Si(R3)2-R4}s-Si(R3)d{[(OSi(R3)2)]f-R3}e(7)式(7)中,R3表示可被取代、并且、1个或1个以上的选自氧、氮、硫和磷中的原子可介于其间的碳原子数1~30的烷基、烷氧基、芳基、或芳烷基,R4表示碳原子数1~10的2价烃基。作为碳原子数1~30的烷氧基,优选碳原子数1~10的烷氧基,作为其具体例,可列举出甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、仲丁氧基、叔丁氧基、正戊氧基、正己氧基、正庚氧基、正辛氧基、正壬氧基、正癸氧基等。此外,作为烷基、芳基、芳烷基,可列举出与上述R1中例示的基团同样的基团。作为上述取代基的具体例,可列举出氟原子、氯原子等上述的各种卤素原子、甲氧基、乙氧基、丙氧基等烷氧基、二烷基氨基等氨基等。作为R4中的碳原子数1~10的2价烃基,可列举出亚乙基、亚丙基等亚烷基,优选为亚乙基。上述s表示0或1的整数,d表示0~3的整数,e表示0~3的整数,并且d+e满足为3,f表示1~300的整数,特别地,优选由s为0的式(7′)所示的甲硅烷基或聚有机硅氧烷基。-Si(R3)d{[(OSi(R3)2)]f-R3}e(7′)作为由式(7)表示的甲硅烷基或聚有机硅氧烷基的具体例,可列举出三甲基甲硅烷基、三乙基甲硅烷基、苯基二甲基甲硅烷基、三甲氧基甲硅烷基、三乙氧基甲硅烷基、五甲基二甲硅烷氧基、双(三甲基甲硅烷氧基)甲基甲硅烷基、三(三甲基甲硅烷氧基)甲硅烷基、-Si(Me)2{OSi(Me)2}f-1-OSiMe3(f与上述相同。)所示的聚二甲基甲硅烷氧基、-Si(Me)2{OSi(Me)2}f-1-OSiMe2nBu(f与上述相同。)所示的聚二甲基甲硅烷氧基等,但并不限定于这些。再有,作为R2,除了由通式(7)表示的基团以外,也能够使用经由硅亚乙基高度地分支的树枝状大分子型的硅氧烷基。这些中,作为R1,优选可被卤素原子取代的碳原子数1~30的1价烃基,优选可被卤素原子取代的碳原子数1~10的烷基,更优选可被卤素原子取代的碳原子数1~5的烷基。式(1)中,a为1或2,b表示0~6的整数,c表示0~3的整数,根据金属元素(M)的价数选择它们,a为1时,b+c满足为2或3,a为2时,b+c满足4~6。优选地,b为2~4。更具体地,式(1)中的M为Fe、Co或Ni的情况下,优选a为1,b为2或0,c为0、2或3,更优选a为1,b为2,c为0。式(1)中的M为Rh的情况下,优选a为2,b为4,c为0。式(1)中的M为Ru的情况下,优选a为2,b为4,c为1。本发明中,作为能够优选作为催化剂前体使用的金属化合物的具体例,可列举出醋酸铁(II)、新戊酸铁(II)、三氟醋酸铁(II)(四氢呋喃络合物、以下THF)、由[Fe(基)(μ-基)]2与各种醇、羧酸或含有硅氧烷的羧酸酯制备的具有铁-氧键的铁络合物等铁化合物;醋酸钴(II)、氯化钴(II)、溴化钴(II)、异丙醇钴(II)、新戊酸钴(II)、三氟醋酸钴(II)(THF)等钴化合物;醋酸镍(II)、新戊酸镍(II)等镍化合物;Ru2(μ-OAc)4Cl等钌化合物;醋酸铑(II)二聚体等铑化合物等,但并不限定于这些。再有,这些金属盐能够作为市售品、或者采用公知文献(J.Cluster.Sci.,2005,16,331.Inorganicchemistry,2007,46,3378.Organometallics,1993,12,2414.Russ.Chem.Bull.,1999,48,1751.J.Inorg.Nucl.Chem.,1966,28,2285.等)记载的方法合成而得到。另一方面,由式(2)表示的配体为1个或2个氮原子邻接的卡宾化合物。式(2)中,Y表示碳原子、氮原子或氧原子,Y为碳原子时,g为3,Y为氮原子时,g为2,Y为氧原子时,g为1。R4和R5相互独立地表示可被卤素原子或烷氧基取代的、碳原子数1~30的烷基、芳基或芳烷基,R4的任一个与R5的任一个可结合构成2价的有机基团而形成环状结构,这种情况下,在环状结构内可含有氮原子和/或不饱和键。碳原子数1~30的烷基、芳基、和芳烷基、以及烷氧基的具体例可列举出与上述例示的基团同样的基团。优选为由式(8)表示的环状的卡宾化合物。[化4]其中,Z为可含有氮原子和/或不饱和键的、碳原子数2~5的2价有机基团,作为其具体例,除了在上述碳原子数1~30的2价有机基团中例示的基团中碳原子数2~5的基团以外,可列举出亚乙烯基、丙-1-烯-1,3-二基(亚丙烯基)等。作为环状卡宾化合物的具体例,可以列举出以下的化合物,但并不限定于这些。[化5]另外,除此以外,也能够使作为前体的咪唑鎓盐与KOtBu这样的碱反应,在体系内边使卡宾化合物产生边进行氢化硅烷化反应。作为前体即咪唑鎓盐的具体例,可以列举出以下的化合物,但并不限定于这些。[化6]再有,本发明的氢化硅烷化反应催化剂中,在不损害其活性等的范围内,可将公知的2电子给予性配体并用。作为2电子给予性配体,并无特别限定,优选羰基以外的配体,可列举出氨分子、醚化合物、胺化合物、膦化合物、亚磷酸酯化合物、硫醚化合物等。制备本发明的氢化硅烷化反应催化剂时,对金属化合物与卡宾化合物的使用量并无特别限定,相对于1当量的金属化合物,优选使卡宾化合物为0.5~10当量左右,更优选1~6当量,进一步优选2~4当量。另外,使用本发明的氢化硅烷化反应催化剂进行氢化硅烷化反应时,对催化剂的使用量并无特别限定,如果考虑在室温~100℃左右的温和的条件下进行反应、高收率地得到目标物,相对于1摩尔作为基质的含有脂肪族不饱和基的化合物,作为金属化合物,优选使用0.1摩尔%以上,更优选使用0.5摩尔%以上。再有,对金属化合物的使用量并无特别的上限,从经济的观点出发,相对于1摩尔基质,为10摩尔%左右,优选为5摩尔%。本发明的氢化硅烷化反应催化剂可以在将由金属化合物和卡宾化合物制备的金属络合物催化剂离析后使用,也可不将其离析,在使具有脂肪族不饱和键的化合物与具有Si-H基的氢硅烷化合物或有机氢聚硅氧烷化合物进行氢化硅烷化反应的体系内由金属化合物和卡宾化合物制备而使用,从操作的简便化等方面出发,优选不离析地在体系内制备而使用。此时,一旦由金属化合物和卡宾化合物制备了催化剂后,可以加入具有脂肪族不饱和键的化合物和具有Si-H基的氢硅烷化合物或有机氢聚硅氧烷化合物,也可平均分为几种成分而进料,还可将全部成分一齐进料。对金属化合物与卡宾化合物的反应条件并无特别限定,通常,反应温度为10~100℃左右,优选为30~80℃,反应时间为1~48小时左右。在催化剂制备时和氢化硅烷化反应时也能够使用有机溶剂,在本发明中优选在无溶剂下进行。使用有机溶剂的情况下,作为其种类,只要不对反应产生影响,则为任意的有机溶剂,例如能够使用戊烷、己烷、庚烷、辛烷、环己烷等脂肪族烃类;乙醚、二异丙醚、二丁醚、环戊基甲基醚、四氢呋喃、1,4-二噁烷等醚类;苯、甲苯、二甲苯、均三甲苯等芳香族烃类等。在使用本发明的氢化硅烷化反应催化剂的氢化硅烷化反应中,只要是含有脂肪族不饱和键的、烯烃化合物、硅烷化合物或有机聚硅氧烷化合物等具有脂肪族不饱和键的化合物与具有Si-H键的、硅烷化合物或有机聚硅氧烷化合物的组合,则能够对这些各化合物的结构无任何限制地使用。另外,使用本发明的氢化硅烷化反应催化剂的氢化硅烷化反应,除了由具有脂肪族不饱和键的烯烃化合物和具有Si-H键的硅烷化合物得到的硅烷偶联剂、由具有脂肪族不饱和键的烯烃化合物和具有Si-H键的有机聚硅氧烷得到的改性硅油类等以外,也能够在由具有脂肪族不饱和键的有机聚硅氧烷化合物和具有Si-H键的有机聚硅氧烷得到的有机硅固化物等以往的使用铂催化剂工业上进行的所有的用途中使用。实施例以下列举合成例和实施例,对本发明更具体地说明,但本发明并不受下述的实施例限制。络合物的合成使用施嫩克技术或手套箱,在氮或氩气氛下进行全部的操作,在金属化合物的制备中使用的溶剂全部在采用公知的方法进行了脱氧、脱水后使用。得到的金属化合物在25℃、氮气气氛下保存,在反应中使用。烯烃的氢化硅烷化反应和溶剂精制全部在非活性气体气氛下进行,各种反应中使用的溶剂等全部使用预先采用公知的方法进行了精制、干燥、脱氧的产物。1H、13C-NMR的测定使用日本电子(株)制造JNM-ECA600、JNM-LA400进行,IR测定使用日本分光(株)制造FT/IR-550进行,元素分析使用PerkinElmer制造2400II/CHN进行,X射线结晶结构解析使用RigakuCorp.((株)リガク)制造的VariMax、MoKα线0.71069埃进行。再有,以下所示的化学结构式中按照惯用的表示法省略了氢原子。另外,OAc是指醋酸阴离子,iPr是指异丙基,NHC是指N-杂环卡宾配体。(1)金属化合物的合成[合成例1]新戊酸铁的合成以文献J.Cluster.Sci.,2005,16,331.作为参考、采用以下的方法合成。在带有回流管的50mL二口茄形烧瓶中加入还原铁0.86g(15.4mmol)、新戊酸3.50g(34.3mmol),在160℃下搅拌了12小时。此时,反应溶液从无色透明变化为绿色。进而,加入新戊酸2.50g(24.5mmol),在160℃下搅拌了19小时。然后,将反应溶液过滤,与回收的上清液合并,在80℃下减压干燥。将得到的固体用己烷清洗,得到了绿色固体(2.66g、收率67%)。FT-IR(KBr)ν:2963,2930,2868,1583,1523,1485,1457,1427,1379,1362,1229,1031,938,900,790,608,576,457cm-1[合成例2]使用了[Fe(基)(μ-基)]2的具有铁-氧键的铁前体的制备以文献Organometallics,1993,12,2414.作为参考、采用下述方法合成。在50mL二口茄形烧瓶中加入镁条1.08g(44.3mmol)、THF35mL,向其中缓慢地滴加溴代均三甲苯8.49g(42.6mmol)。滴加后,确认发生了放热后,在60℃下搅拌了3小时。用玻璃过滤器将得到的溶液过滤,制备了溴代基镁格利雅试剂的THF溶液。在100mL施嫩克管中加入FeCl22.63g(20.7mmol)、THF30mL、1,4-二噁烷10mL,冷却到-78℃。在其中缓慢地加入上述制备的溴代基镁格利雅试剂的THF溶液,在25℃下搅拌了2小时。此时,反应液从茶色悬浊液变为红色悬浊液。然后,通过离心分离去除析出的固体,减压干燥。将得到的红色固体溶解于乙醚中,再次通过离心分离将固体除去后,在-30℃下使其重结晶而得到了结晶(4.36g、收率72%)。对于得到的结晶,在C6D6中测定1H-NMR,进行鉴定。1H-NMR(600MHz,C6D6)δ:23.68(s,2H),23.17(s,2H),21.44(s,3H),17.94(s,3H),10.19(s,6H),-6.66(s,6H)。在20mL施嫩克管中加入上述得到的[Fe(基)(μ-基)]23mg(0.01mmol)或9mg(0.015mmol),在THF1mL中溶解。在其中加入1,1,1,3,3,3-六氟异丙醇16mg(0.09mmol),在25℃下搅拌了30分钟。然后,减压干燥,制备了具有铁-氧键的铁前体(Fe(OR)2)。[合成例3]新戊酸钴的合成以文献Russ.Chem.Bull.,1999,48,1751.作为参考、采用以下的方法合成。在带有回流管的50mL二口茄形烧瓶中加入醋酸钴1.15g(6.5mmol)、新戊酸1.55g(15.2mmol)、新戊酸酐0.5mL(2.5mmol),在160℃下搅拌了1小时。此时,反应溶液从浅紫色变为紫色。然后,在80℃下减压干燥,将得到的固体用戊烷和乙醚清洗,干燥,得到了紫色固体(1.15g、收率68%)。FT-IR(KBr)ν:2963,2929,2868,1599,1524,1485,1457,1420,1379,1363,1229,1032,938,900,792,613,585,460cm-1[合成例4]Ru2(μ-OAc)4Cl的合成以文献J.Inorg.Nucl.Chem.,1966,28,2285作为参考、采用以下的方法合成。在200mL二口茄形烧瓶中加入RuCl33水合物1.09g(4.18mmol)、冰醋酸35mL、醋酸酐7mL,在145℃下搅拌了2小时。冷却后,暂且过滤,再次在145℃下搅拌了6小时。然后,在-30℃下使其结晶,用冰醋酸、甲醇、乙醚清洗,得到了红茶色的固体(61mg、收率6%)。FT-IR(KBr)ν:3023,2991,2934,1643,1444,1401,1356,1041,1015,944,691,625,606cm-1[合成例5]钴络合物A的合成在20mL施嫩克管中加入合成例3中得到的新戊酸钴0.20g(0.77mmol)、1,3-双(2,6-二异丙基苯基)咪唑-2-亚基(以下记为IPr)0.60g(1.53mmol)、甲苯20mL,在25℃下搅拌了12小时。然后,在减压下将溶剂馏除,用乙醚/己烷混合溶液(10mL/10mL)萃取后,再次在减压下浓缩到约5mL。使该溶液在-30℃下重结晶,得到了透明结晶(0.20g、收率40%)。将得到的钴络合物A的X射线结晶结构解析结果示于图1中,将1H-NMR波谱示于图2中。FT-IR(KBr)ν:3158,3123,3077,2962,2924,2871,1586,1565[ν(COCtBu3-κ2)],1549,1530,1482,1463,1447,1413,1358,1331,1258,1222,1181,1117,1100,1061,1028,947,938,900,807,793,752,695,610,546,536cm-11H-NMR(600MHz、CDCl3)δ:-15.59(s,12H),2.54(br,4H),3.78(s,12H),6.16(br,4H),6.55(br,2H),15.64(s,18H),53.88(s,2H)。[合成例6]铁络合物B的合成在20mL施嫩克管中加入合成例1中得到的新戊酸铁0.10g(0.39mmol)、IPr0.18g(0.46mmol)、甲苯10mL,在25℃下搅拌了12小时。然后,添加己烷,萃取后,再次在减压下浓缩到约5mL。使该溶液在-30℃下重结晶,得到了透明结晶(0.06g、收率26%)。将得到的铁络合物B的X射线结晶结构解析结果示于图3中,将1H-NMR波谱示于图4中。1H-NMR(600MHz,C6D6)δ:-38.68(br,4H),-7.07(br,12H),-4.05(s,12H),22.99(s,4H),24.32(s,2H),26.18(s,2H),33.68(s,18H)。(1)使用了具有金属-氧键的络合物和NHC配体的1-辛烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化[化7][实施例1]使用了新戊酸铁和1,3-二基咪唑-2-亚基(以下记为IMes)的氢化硅烷化在螺纹瓶中加入作为催化剂前体的合成例1中得到的新戊酸铁8mg(0.03mmol)、作为NHC配体的IMes18mg(0.06mmol)、1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、1-辛烯157μL(1.0mmol),密闭后在80℃下搅拌了24小时。然后,将成为内部标准的茴香醚1.0mmol加入反应溶液中,搅拌后,使极少量溶解于重氯仿中,通过氧化铝将催化剂除去,进行了1H-NMR测定。以后所示的实施例同样地按照该程序制备测定用样品,测定了1H-NMR波谱。其结果确认了成为原料的1-辛烯的乙烯部位的信号完全地消失了。而且,确认目标生成物1,1,1,3,3-五甲基-3-辛基二硅氧烷中的与硅邻接的碳上的质子的信号即0.51ppm的多重线,求出了其收率。将结果示于表1中。基于1H-NMR的收率为65%。[实施例2]使用了具有铁-氧键的铁络合物和IMes的氢化硅烷化首先,按照合成例2中所示的程序,由[Fe(基)(μ-基)]29mg(0.015mmol)和1,1,1,3,3,3-六氟异丙醇16mg(0.09mmol)制备了具有铁-氧键的铁前体(Fe(OR)2)。在其中加入IMes18mg(0.06mmol)、1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、1-辛烯157μL(1.0mmol),密闭后在80℃下搅拌了24小时。冷却后,测定1H-NMR波谱,确定了生成物的结构和收率。其结果确认了原料的信号完全地消失了。而且,确认目标生成物的信号即0.51ppm的多重线,求出了其收率。基于1H-NMR的收率为15%。[实施例3]实施例1的金属化合物改变除了代替新戊酸铁而使用了醋酸钴5mg(市售品、0.03mmol)以外,按照与实施例1同样的程序进行了反应。其结果确认了原料的信号完全地消失了。而且,确认目标生成物的信号即0.51ppm的多重线,求出了其收率。将结果示于表1中。基于1H-NMR的收率为78%。[实施例4]实施例3的配体改变除了代替IMes而使用了IPr23mg(0.06mmol)以外,按照与实施例3同样的程序进行了反应。其结果确认了原料的信号完全地消失了。而且,确认目标生成物的信号即0.51ppm的多重线,求出了其收率。将结果示于表1中。基于1H-NMR的收率为99%以上。[实施例5]实施例3的配体改变除了代替IMes而使用了1,3-双(2,6-二异丙基苯基)咪唑啉-2-亚基23mg(0.06mmol)以外,按照与实施例3同样的程序进行了反应。其结果确认了原料的信号完全地消失了。而且,确认目标生成物的信号即0.51ppm的多重线,求出了其收率。将结果示于表1中。基于1H-NMR的收率为58%。[实施例6]实施例3的配体改变除了代替IMes而使用了1,3-二-叔丁基咪唑-2-亚基11mg(0.06mmol)以外,按照与实施例3同样的程序进行了反应。其结果确认了原料的信号减少了。而且,确认目标生成物的信号即0.51ppm的多重线,求出了其收率。将结果示于表1中。基于1H-NMR的转化率为60%,收率为19%。[实施例7]实施例3的金属化合物改变除了代替醋酸钴而使用了合成例3中得到的新戊酸钴8mg(0.03mmol)以外,按照与实施例3同样的程序进行了反应。其结果确认了原料的信号完全地消失了。而且,确认目标生成物的信号即0.51ppm的多重线,求出了其收率。将结果示于表1中。基于1H-NMR的收率为99%以上。[实施例8]实施例6的金属化合物改变除了代替醋酸钴而使用了醋酸铑二聚体7mg(市售品、0.015mmol)以外,按照与实施例6同样的程序进行了反应。其结果确认了成为原料的1-辛烯的乙烯部位的信号完全地消失了以及目标生成物1,1,1,3,3-五甲基-3-辛基二硅氧烷中的与硅邻接的碳上的质子的信号即0.51ppm的多重线。基于1H-NMR的收率为99%以上。[实施例9]使用了空气中保存的新戊酸钴和IMes的氢化硅烷化在装入了搅拌子的螺纹瓶中使合成例3中得到的新戊酸钴8mg(0.03mmol)在空气中(25℃、60%RH)暴露1天,然后加入了IMes18mg(0.06mmol)、1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、1-辛烯157μL(1.0mmol)。将该容器内用氮置换后,在80℃下搅拌了24小时。冷却后,测定1H-NMR波谱,确定了生成物的结构和收率。其结果,确认了原料的信号完全地消失了。而且,确认目标生成物的信号即0.51ppm的多重线,求出其收率。将结果示于表1中。基于1H-NMR的收率为99%以上。[表1](2)使用了具有金属-氧键的络合物和NHC配体的各种烯烃的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化[实施例10]使用了新戊酸钴和IMes的烯丙基苯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化在反应容器中加入合成例3中得到的新戊酸钴8mg(0.03mmol)、IMes18mg(0.06mmol)、1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、烯丙基苯133μL(1.0mmol),在80℃下搅拌了24小时。冷却后,采用内部标准法,可知获得基质转化率99%以上,以收率30%得到1,1,1,3,3-五甲基-3-(3-苯基丙基)二硅氧烷,以收率33%得到了丙基苯。[实施例11]使用了新戊酸钴和IMes的乙烯基三甲基硅烷的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化在反应容器中加入合成例3中得到的新戊酸钴8mg(0.03mmol)、IMes18mg(0.06mmol)、1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、乙烯基三甲基硅烷145μL(1.0mmol),在80℃下搅拌了24小时。冷却后,采用内部标准法,可知获得基质转化率99%以上,以收率20%得到了1,1,1,3,3-五甲基-3-(2-三甲基甲硅烷基乙基)二硅氧烷,以收率15%得到了作为脱氢化生成物的1,1,1,3,3-五甲基-3-(2-三甲基甲硅烷基乙烯基)二硅氧烷。[实施例12]使用了新戊酸钴和IMes的2-降冰片烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化在反应容器中加入合成例3中得到的新戊酸钴8mg(0.03mmol)、IMes18mg(0.06mmol)、1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、2-降冰片烯94mg(1.0mmol),在80℃下搅拌了24小时。冷却后,采用内部标准法,可知获得了基质转化率99%以上,以收率80%得到了1-(1,1,3,3,3-五甲基二硅氧烷基)-2-降冰片烯。[实施例13]使用了新戊酸钴和IMes的1,7-辛二烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化在反应容器中加入合成例3中得到的新戊酸钴8mg(0.03mmol)、IMes18mg(0.06mmol)、1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、1,7-辛二烯151μL(1.0mmol),在80℃下搅拌了24小时。冷却后,采用内部标准法,可知获得基质转化率99%以上,以收率60%得到1,8-双(1,1,3,3,3-五甲基二硅氧烷基)辛烷,以收率5%得到了作为异构化物的辛二烯。[实施例14]使用了新戊酸铁和IMes的1,7-辛二烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化在反应容器中加入合成例1中得到的新戊酸铁8mg(0.03mmol)、IMes18mg(0.06mmol)、1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、1,7-辛二烯151μL(1.0mmol),在80℃下搅拌了24小时。冷却后,采用内部标准法,可知获得了基质转化率99%以上,以收率60%得到了1,8-双(1,1,3,3,3-五甲基二硅氧烷基)辛烷,以收率40%得到了将一者氢化而得到的辛烯。[实施例15]使用了新戊酸钴和IMes的2-辛烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化在反应容器中加入合成例3中得到的新戊酸钴8mg(0.03mmol)、IMes18mg(0.06mmol)、1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、2-辛烯157μL(1.0mmol),密闭后,在80℃下搅拌了24小时。冷却后,采用内部标准法,获得了基质转化率99%以上,以收率83%得到了1,1,1,3,3-五甲基-3-辛基二硅氧烷。(3)使用了金属络合物的氢化硅烷化[实施例16]使用了钴络合物A的1-辛烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化在反应容器中加入合成例5中得到的钴络合物A3mg(0.005mmol)、1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、1-辛烯157μL(1.0mmol),在80℃下搅拌了24小时。冷却后,采用内部标准法,可知基质转化率为99%以上,以收率65%得到了1,1,1,3,3-五甲基-3-辛基二硅氧烷,以收率10%得到了作为异构化物的内部辛烯。[实施例17]使用了钴络合物A的2-降冰片烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化在反应容器中加入合成例5中得到的钴络合物A6mg(0.01mmol)、1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、2-降冰片烯94mg(1.0mmol),在80℃下搅拌了24小时。冷却后,采用内部标准法,可知基质转化率为58%,以收率26%得到了1-(双环[2.2.1]庚-2-基)-1,1,3,3,3-五甲基二硅氧烷,以收率1%得到了降冰片烷。[实施例18]使用了钴络合物A的2-降冰片烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化在反应容器中加入合成例5中得到的钴络合物A6mg(0.01mmol)、IPr4mg(0.01mmol)、1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、2-降冰片烯94mg(1.0mmol),在80℃下搅拌了24小时。冷却后,采用内部标准法,可知基质转化率为40%,以收率11%得到了1-(双环[2.2.1]庚-2-基)-1,1,3,3,3-五甲基二硅氧烷,以收率1%得到了降冰片烷。[实施例19]使用了铁络合物B的1-辛烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化在反应容器中加入合成例6中得到的铁络合物B6mg(0.01mmol)、1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、1-辛烯157μL(1.0mmol),在80℃下搅拌了24小时。冷却后,采用内部标准法,可知基质转化率为>99%,以收率5%得到了1,1,1,3,3-五甲基-3-辛基二硅氧烷,以收率84%得到了作为1-辛烯异构化的生成物的内部辛烯。[实施例20]使用了铁络合物B的1-辛烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化在反应容器中加入合成例6中得到的铁络合物B6mg(0.01mmol)、IPr4mg(0.01mmol)、1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、1-辛烯157μL(1.0mmol),在80℃下搅拌了24小时。冷却后,采用内部标准法,可知基质转化率为>99%,以收率8%得到了1,1,1,3,3-五甲基-3-辛基二硅氧烷,以收率82%得到了作为1-辛烯异构化的生成物的内部辛烯。(4)使用了新戊酸铁和NHC配体的2-降冰片烯的采用各种氢硅烷的氢化硅烷化反应[化8][实施例21]使用了新戊酸铁和IMes的2-降冰片烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化反应在螺纹瓶中加入作为催化剂的合成例1中得到的新戊酸铁3mg(0.01mmol)、作为NHC配体的IMes6mg(0.02mmol)、作为氢硅烷的1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、2-降冰片烯94mg(1.0mmol),在80℃下搅拌了24小时。冷却后,测定1H-NMR波谱,确定了生成物的结构和收率。其结果,确认了原料的信号完全地消失了。而且,确认目标生成物中的与硅邻接的碳上的质子的信号即0.49ppm的多重线,求出了其收率。将其结果示于表2中。[实施例22]使用了新戊酸铁和IMes的2-降冰片烯的采用1,1,1,3,5,5,5-七甲基三硅氧烷的氢化硅烷化反应在螺纹瓶中加入作为催化剂的合成例1中得到的新戊酸铁3mg(0.01mmol)、作为NHC配体的IMes6mg(0.02mmol)、作为氢硅烷的1,1,1,3,5,5,5-七甲基三硅氧烷353μL(1.3mmol)、2-降冰片烯94mg(1.0mmol),在80℃下搅拌了24小时。冷却后,测定1H-NMR波谱,确定了生成物的结构和收率。其结果确认了原料的信号减少了。而且,确认目标生成物中的与硅邻接的碳上的质子的信号即0.45ppm的多重线,求出其收率。将其结果示于表2。[实施例23]使用了新戊酸铁和IPr的2-降冰片烯的采用三甲氧基硅烷的氢化硅烷化在螺纹瓶中加入作为催化剂的合成例1中得到的新戊酸铁3mg(0.01mmol)、作为NHC配体的IPr8mg(0.02mmol)、作为氢硅烷的三甲氧基硅烷165μL(1.3mmol)、2-降冰片烯94mg(1.0mmol),在80℃下搅拌了24小时。冷却后,测定1H-NMR波谱,确定了生成物的结构和收率。其结果,确认了原料的信号减少了。而且,确认目标生成物中的与硅邻接的碳上的质子的信号即0.76ppm的多重线,求出其收率。将其结果示于表2中。[表2](5)使用了新戊酸钴和NHC配体的1-辛烯的采用各种氢硅烷的氢化硅烷化反应[化9][实施例24]使用了新戊酸钴和IMes的1-辛烯的采用1,1,1,3,5,5,5-七甲基三硅氧烷的氢化硅烷化反应在螺纹瓶中加入作为催化剂的合成例3中得到的新戊酸钴3mg(0.01mmol)、作为NHC配体的IMes6mg(0.02mmol)、作为氢硅烷的1,1,1,3,5,5,5-七甲基三硅氧烷353μL(1.3mmol)、1-辛烯157μL(1.0mmol),在80℃下搅拌了24小时。冷却后,测定了1H-NMR波谱,确定了生成物的结构和收率。其结果确认了原料的信号完全地消失了。而且,确认目标生成物中的与硅邻接的碳上的质子的信号即0.49ppm的多重线,求出了其收率。将其结果示于表3中。[实施例25]使用了IPr作为配体的实施例20的氢化硅烷化反应除了作为配体代替IMes而使用了IPr8mg(0.02mmol)以外,按照与实施例20同样的程序进行了反应。其结果确认了原料的信号减少。而且,确认目标生成物中的与硅邻接的碳上的质子的信号即0.49ppm的多重线,求出了其收率。将其结果示于表3中。[实施例26]使用了新戊酸钴和IMes的1-辛烯的采用乙基二甲基硅烷的氢化硅烷化反应在螺纹瓶中加入作为催化剂的合成例3中得到的新戊酸钴3mg(0.01mmol)、作为NHC配体的IMes6mg(0.02mmol)、作为氢硅烷的乙基二甲基硅烷165μL(1.3mmol)、1-辛烯157μL(1.0mmol),在80℃下搅拌了24小时。冷却后,测定1H-NMR波谱,确定了生成物的结构和收率。其结果确认了原料的信号完全地消失了。而且,确认目标生成物中的与硅邻接的碳上的质子的信号即0.47ppm的多重线,求出了其收率。将其结果示于表3中。[实施例27]使用了新戊酸钴和IMes的1-辛烯的采用三甲氧基硅烷的氢化硅烷化反应在螺纹瓶中加入作为催化剂的合成例3中得到的新戊酸钴3mg(0.01mmol)、作为NHC配体的IMes6mg(0.02mmol)、作为氢硅烷的三甲氧基硅烷171μL(1.3mmol)、1-辛烯157μL(1.0mmol),在80℃下搅拌了24小时。冷却后,测定1H-NMR波谱,确定了生成物的结构和收率。其结果确认了原料的信号完全地消失。而且,确认目标生成物中的与硅邻接的碳上的质子的信号即0.65ppm的多重线,求出了其收率。将其结果示于表3中。[表3](3)使用了新戊酸钴和NHC配体的2-降冰片烯的采用各种氢硅烷的氢化硅烷化反应[化10][实施例28]使用了新戊酸钴和IMes的2-降冰片烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化反应在螺纹瓶中加入作为催化剂的合成例3中得到的新戊酸钴3mg(0.01mmol)、作为NHC配体的IMes6mg(0.02mmol)、作为氢硅烷的1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、2-降冰片烯94mg(1.0mmol),在80℃下搅拌了24小时。冷却后,测定1H-NMR波谱,确定了生成物的结构和收率。其结果确认了原料的信号完全地消失了。而且,确认目标生成物中的与硅邻接的碳上的质子的信号即0.49ppm的多重线,求出了其收率。将其结果示于表4中。[实施例29]使用了新戊酸钴和IMes的2-降冰片烯的采用二甲基苯基硅烷的氢化硅烷化反应在螺纹瓶中加入作为催化剂的合成例3中得到的新戊酸钴3mg(0.01mmol)、作为NHC配体的IMes6mg(0.02mmol)、作为氢硅烷的二甲基苯基硅烷202μL(1.3mmol)、2-降冰片烯94mg(1.0mmol),在80℃下搅拌了24小时。冷却后,测定1H-NMR波谱,确定了生成物的结构和收率。其结果确认了原料的信号完全地消失了。而且,确认目标生成物中的与硅邻接的碳上的质子的信号即0.81ppm的多重线,求出了其收率。将其结果示于表4中。[实施例30]使用了IPr作为配体的实施例29的氢化硅烷化反应除了作为配体代替IMes而使用了IPr8mg(0.02mmol)以外,按照与实施例25同样的程序进行了反应。其结果确认了原料的信号减少了。而且,确认目标生成物中的与硅邻接的碳上的质子的信号即0.81ppm的多重线,求出了其收率。将其结果示于表4中。[实施例31]使用了新戊酸钴和IMes的2-降冰片烯的采用三甲氧基硅烷的氢化硅烷化反应在螺纹瓶中加入作为催化剂的合成例3中得到的新戊酸钴3mg(0.01mmol)、作为NHC配体的IMes6mg(0.02mmol)、作为氢硅烷的三甲氧基硅烷165μL(1.3mmol)、2-降冰片烯94mg(1.0mmol),在80℃下搅拌了24小时。冷却后,测定1H-NMR波谱,确定了生成物的结构和收率。其结果确认了原料的信号完全地消失了。而且,确认目标生成物中的与硅邻接的碳上的质子的信号即0.76ppm的多重线,求出了其收率。将其结果示于表4中。[实施例32]使用了IPr作为配体的实施例31的氢化硅烷化反应除了作为配体代替IMes而使用了IPr8mg(0.02mmol)以外,按照与实施例27同样的程序进行了反应。其结果确认了原料的信号减少了。而且,确认目标生成物中的与硅邻接的碳上的质子的信号即0.76ppm的多重线,求出了其收率。将其结果示于表4中。[表4][合成例7]钴羧酸盐C的合成在带有回流管的1L的烧瓶中装入10-十一碳烯酸184.0g(1.0mol)、甲苯150.0g,加热到80℃。向其中滴入六甲基二硅氮烷100.6g(0.625mol),滴入结束后,在80℃下进一步加热3小时。在减压下加热到100℃,将挥发成分除去,得到了CH2=CH(CH2)8COOSiMe3(硅烷化物A)(254.4g、收率99.4%)。在带有回流管的1L的烧瓶中装入上述硅烷化物A254.4g(0.99mol)、甲苯100.0g,加热到90℃。向其中加入氯铂酸0.5质量%的甲苯溶液0.5g,滴入1,1,1,3,5,5,5-七甲基三硅氧烷264.7g(1.19mol)。滴入结束后,在100℃下进一步加热了2小时。在减压下加热到120℃,将挥发成分除去,得到了(Me3SiO)2MeSi(CH2)10COOSiMe3(加成物A)(451.2g、收率95.0%)。在1L的烧瓶中装入加成物A239.0g(0.5mol)、甲醇140.0g,在室温下搅拌了14小时。蒸馏,得到了目标物(Me3SiO)2MeSi(CH2)10COOH(沸点175.0~176.0℃/0.3kPa、产量162.4g、收率80.0%)。基于气相色谱的纯度为99.5%。在20mL茄形烧瓶中加入醋酸钴0.43g(2.41mmol)、(Me3SiO)2MeSi(CH2)10COOH2.0g(4.92mmol),在180℃下搅拌了1小时。然后,在这样的温度下减压干燥1小时,制备了钴羧酸盐C。将钴羧酸盐C的FT-IR光谱示于图5中。FT-IR(KBr)ν:2958,2924,2583,1555,1413,1257,1078,1049,842,799,783,754,687.(4)使用了钴羧酸盐C和IMes的1-辛烯的采用各种硅烷的氢化硅烷化反应[化11][实施例33]使用了钴羧酸盐C和IMes的1-辛烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化反应在螺纹瓶中加入作为催化剂的合成例7中得到的钴羧酸盐C9mg(0.01mmol)、作为NHC配体的IMes6mg(0.02mmol)、作为氢硅烷的1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、1-辛烯157μL(1.0mmol),在80℃下搅拌了24小时。冷却后,测定1H-NMR波谱,确定了生成物的结构和收率。其结果确认了原料的信号消失了。而且,确认目标生成物中的与硅邻接的碳上的质子的信号即0.50ppm附近的多重线,求出了其收率。将其结果示于表5中。[实施例34]使用了钴羧酸盐C和IMes的1-辛烯的采用两末端氢二甲基甲硅烷氧基封端聚二甲基硅氧烷的氢化硅烷化反应在螺纹瓶中加入作为催化剂的合成例7中得到的钴羧酸盐C9mg(0.01mmol)、作为NHC配体的IMes6mg(0.02mmol)、作为氢硅烷的两末端氢二甲基甲硅烷氧基封端聚二甲基硅氧烷(聚合度27)1.07g(0.50mmol)、1-辛烯157μL(1.0mmol),在80℃下搅拌了24小时。冷却后,测定1H-NMR波谱,确定了生成物的结构和收率。其结果确认了原料的信号消失了。而且,确认目标生成物中的与硅邻接的碳上的质子的信号即0.50ppm附近的多重线,求出了其收率。将其结果示于表5中。[表5](5)使用了新戊酸钴和IMes的1-辛烯的采用氢硅烷两末端聚二甲基硅氧烷的氢化硅烷化反应[化12][实施例35]在螺纹瓶中加入作为催化剂的合成例3中得到的新戊酸钴8mg(0.03mmol)、作为NHC配体的IMes18mg(0.06mmol)、作为氢硅烷的氢硅烷两末端聚二甲基硅氧烷(聚合度27)1.39g(0.65mmol)、1-辛烯157μL(1.0mmol),在80℃下搅拌了24小时。冷却后,测定1H-NMR波谱,确定了生成物的结构和收率。其结果确认了原料的信号消失了。而且,确认目标生成物中的与硅邻接的碳上的质子的信号即0.65ppm附近的多重线,求出了其收率。将其结果示于表6中。[表6]金属盐转化率(%)收率(%)实施例35新戊酸钴>9985[合成例8]钴羧酸盐D的合成在带有回流管的500mL的烧瓶中装入3-丁烯酸100.0g(1.16mol)、己烷80.0g,加热到70℃。向其中滴入六甲基二硅氮烷117.0g(0.73mol),滴入结束后,在70℃下进一步加热了3小时。对反应液进行蒸馏,得到了目标物CH2=CHCH2COOSiMe3(硅烷化物B)(沸点60.0~62.0℃/5.3kPa、产量155.1g、收率84.6%)。基于气相色谱的纯度为94.4%。接下来,在带有回流管的500mL的烧瓶中装入上述硅烷化物B155.1g(0.98mol)、甲苯150.0g,加热到90℃。在其中加入氯铂酸0.5质量%的甲苯溶液0.5g,滴入了1,1,1,3,5,5,5-七甲基三硅氧烷239.8g(1.08mol)。滴加结束后,在100℃下进一步加热了2小时。对反应液进行蒸馏,得到了目标物(Me3SiO)2MeSi(CH2)3COOSiMe3(加成物B)(沸点97.0~98.5℃/0.3kPa、产量253.8g、收率68.1%)。基于气相色谱的纯度为98.7%。接下来,在500mL的烧瓶中装入加成物B207.5g(0.55mol)、甲醇100.0g,在室温下搅拌了14小时。进行蒸馏,得到了目标物(Me3SiO)2MeSi(CH2)3COOH(沸点119.5~121.0℃/0.3kPa、产量109.5g、收率64.6%)。基于气相色谱的纯度为98.9%。在20mL茄形烧瓶中加入醋酸钴0.20g(1.13mmol)、(Me3SiO)2MeSi(CH2)3COOH0.70g(2.28mmol),在160℃下搅拌了1小时。然后,在这样的温度下减压干燥1小时,制备了钴羧酸盐D。将钴羧酸盐D的FT-IR光谱示于图6中。FT-IR(KBr)ν:2958,2901,2880,1686,1561,1413,1259,1176,1078,1041,842,797,755.[合成例9]钴羧酸盐E的合成在带有回流管的1L的烧瓶中装入10-十一碳烯酸184.0g(1.0mol)、甲苯150.0g,加热到80℃。向其中滴入六甲基二硅氮烷100.6g(0.625mol),滴入结束后,在80℃下进一步加热了3小时。在减压下加热到100℃,将挥发成分除去,得到了CH2=CH(CH2)8COOSiMe3(与合成例7的硅烷化物A相同)(产量254.3g、收率99.3%)。在带有回流管的1L的烧瓶中装入硅烷化物A51.2g(0.20mol),加热到90℃。在其中加入氯铂酸0.5质量%的甲苯溶液0.2g,滴入nBu(Me2)SiO(Me2SiO)3Si(Me2)H94.5g(0.23mol)。滴加结束后,在100℃下进一步加热了2小时。在减压下加热到200℃,将未反应物除去,得到了目标物nBu(Me2)SiO(Me2SiO)3Si(Me2)(CH2)10COOSiMe3(加成物C)(产量127.0g、收率95.0%)。在500mL的烧瓶中装入加成物C127.0g(0.19mol)、甲醇100.0g,在室温下搅拌了14小时。在减压下加热到100℃,将挥发成分除去,得到了目标物nBu(Me2)SiO(Me2SiO)3Si(Me2)(CH2)10COOH(产量111.0g、收率98.0%)。基于气相色谱的纯度为99.8%。在20mL茄形烧瓶中加入醋酸钴0.20g(1.13mmol)、nBu(Me2SiO)5(CH2)10COOH1.35g(2.26mmol),在160℃下搅拌了1小时。然后,在这样的温度下减压干燥1小时,制备了钴羧酸盐E。将钴羧酸盐E的FT-IR光谱示于图7中。FT-IR(KBr)ν:2960,2924,2854,1560,1457,1412,1259,1088,1037,840,798.(5)使用了钴羧酸盐D或钴羧酸盐E和IMes的1-辛烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化反应[化13][实施例36]使用了钴羧酸盐D和IMes的1-辛烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化反应在螺纹瓶中加入作为催化剂的合成例7中得到的钴羧酸盐D7mg(0.01mmol)、作为NHC配体的IMes6mg(0.02mmol)、作为氢硅烷的1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、1-辛烯157μL(1.0mmol),在80℃下搅拌了24小时。冷却后,测定1H-NMR波谱,确定了生成物的结构和收率。其结果确认了原料的信号消失了。而且,确认目标生成物中的与硅邻接的碳上的质子的信号即0.50ppm附近的多重线,求出了其收率。将其结果示于表7中。[实施例37]使用了钴羧酸盐E和IMes的1-辛烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化反应在螺纹瓶中加入作为催化剂的合成例8中得到的钴羧酸盐E13mg(0.01mmol)、作为NHC配体的IMes6mg(0.02mmol)、作为氢硅烷的1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、1-辛烯157μL(1.0mmol),在80℃下搅拌了24小时。冷却后,测定1H-NMR波谱,确定了生成物的结构和收率。其结果确认了原料的信号消失了。而且,确认目标生成物中的与硅邻接的碳上的质子的信号即0.50ppm附近的多重线,求出了其收率。将其结果示于表7中。[表7]钴盐转化率(%)收率(%)实施例36钴羧酸盐D>9977实施例37钴羧酸盐E>9982(6)使用了具有镍-氧键的络合物和NHC配体的1-辛烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化[实施例38]使用了醋酸镍和IPr的1-辛烯的采用1,1,3,3,3-五甲基二硅氧烷的氢化硅烷化反应在螺纹瓶中加入作为催化剂前体的醋酸镍5mg(0.03mmol)、作为NHC配体的IPr23mg(0.06mmol)、1,1,3,3,3-五甲基二硅氧烷254μL(1.3mmol)、1-辛烯157μL(1.0mmol),在80℃下搅拌了24小时。冷却后,采用内部标准法,可知基质转化率为32%,以收率27%得到了1,1,1,3,3-五甲基-3-辛基二硅氧烷,以收率5%得到了作为1-辛烯异构化的生成物的内部辛烯。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1