一种催化氯化氢氧化的催化剂及其制备方法和应用与流程
本发明属于催化剂领域与环境保护领域,具体涉及一种催化氯化氢氧化的催化剂及其制备方法和应用。
背景技术:
:
氯气是一种重要的化工基础原料,广泛应用于聚氨酯、有机硅、环氧树脂、氯化橡胶、氯化高聚物、氯化烃等新材料行业,应用于多晶硅制造等新能源行业,应用于消毒剂、洗涤剂、食品添加剂、化妆品助剂等生活精细化工行业,应用于合成甘油、氯苯系列、氯乙酸、氯化苄、等农药医药行业,以及应用于造纸、纺织、冶金和石油化工等行业。
在大部分耗氯行业中,在氯气的使用中出现了两个严重的制约因素。如氯碱工业中,每生产一吨氯气的耗电量超高,而且副产的氯化氢或盐酸的出路困难。而氯化氢氧化制造氯气是解决这两个问题的很好思路。就最近几十年的发展来看,催化氧化法是最有效的解决方案,尤其是经由Deacon反应的催化氧化法,由于操作简单、设备成本低的特点,最具工业化潜力。
Deacon反应是在有催化剂的情况下将氯化氢氧化成氯气的反应。催化剂的类型直接影响Deacon反应氯化氢的转化率。目前Deacon反应所使用的催化剂主要采用铬、铜及钌等金属元素。其中,钌系催化剂由于价格昂贵,限制了其广泛应用。铜系催化剂仅在高温下具有较高活性;铬系催化剂虽然具有较好的低温活性,但稳定性差,而且由于六价铬的毒性,对环境污染大。因此,开发更高性能的铜系、铬系催化剂仍是目前的研究重点。
EP0184413中公开了以氧化铬为催化剂催化氧化氯化氢的方法,但催化剂活性较低,氯化氢的转化率在70%左右。CN85109387A公开了一种利用沉淀法合成以氧化铬为主组分的催化剂,虽然活性能够达到80%左右。但是这些催化剂稳定性都不高,因为铬与氯气极易形成低沸点的氯氧化铬(CrO2Cl2)和氢氧化氧铬(CrO2(OH)2)。较高的反应温度或长时间反应容易使催化剂失活。US5707919公开了一种氧化铬催化剂的改进方法,加入少量铜盐、钾盐和稀土金属作为助剂,所制备的催化剂活性高,稳定性有提高,氯化氢转化率达85.2%。CN104785271A公开了一种利用造粒法制备以氧化铬为主要成分催化剂,添加铜、硼、碱金属、稀土金属等助剂,以铝溶胶、硅溶胶作为载体。催化剂催化活性高,稳定性较好,氯化氢的转化率在80%以上,并具有一定的抗粘结性以及较好的机械强度,可以用于流化床反应器。但由于六价铬具有较大毒性,氧化铬催化剂存在的环境污染问题,限制了其在工业上的应用。
综上所述,尽管国内外研究者在改进铬系催化剂方面做出了诸多的努力,但到目前为止,基于铬系催化剂的氯化氢氧化工艺仍未实现工业化。因此,制备高活性、高稳定性、无毒的新型氯化氢催化氧化催化剂,仍具有重要意义。
技术实现要素:
:
为了克服现有技术中的众多不足,本发明旨在提供一种催化活性高、稳定性高、毒性小的催化氯化氢氧化的催化剂。
此外,本发明还提供了所述催化剂的制备方法及其在催化氯化氢氧化中的应用。
一种催化氯化氢氧化的催化剂,包含化学式为MCr2O4的铬酸盐的至少一种;其中,M选自Co或Zn。
本发明中,所述的催化剂包含铬酸钴、铬酸锌中的中的至少一种。本发明的催化剂具有高活性、高稳定性、无毒及成本低的特点,既适用于大规模氯化氢催化氧化制氯气,也适用于工业氯化氢废气的催化氧化转化。
作为优选,所述的催化剂中,所述铬酸盐具有尖晶石结构。
所述的MCr2O4中,M为2价的Co或Zn金属离子。
作为优选,所述的催化剂为铬酸钴。本发明申请人发现,采用铬酸钴催化氯化氢氧化制备氯气的催化性能、稳定性等性能均进一步提升。
作为优选,所述的催化剂中,铬酸盐中,M与Cr的摩尔比为1∶1~1∶4。
进一步优选,所述的催化剂中,铬酸盐中,M与Cr的摩尔比为1∶2~1∶3。
更为优选,所述的催化剂中,铬酸盐中,M与Cr的摩尔比为1∶2。本发明人还发现,在该比例下,所述的催化剂为是尖晶石结构的铬酸盐,HCl的催化氧化活性更强,且催化剂的稳定性也有很大的提升。M过量的时候催化活性会下降,反之,当Cr过量的时候催化剂的稳定性会下降。
本发明人发现,所述的催化剂的优选为比表面积15-30m2/g;晶型为尖晶石型、晶体粒径大小在10~50nm。在该优选条件下,催化剂的稳定性强、催化活性高。
进一步优选,所述的催化剂的优选为比表面积25-30m2/g;晶体粒径大小在15~30nm。
所述的催化剂以所述的铬酸盐为主要活性化合物,铬酸盐含量越高催化活性越高;本发明人发现,所述的催化剂中,催化活性成分铬酸钴和/或铬酸锌的重量百分数大于或等于50%。在该重量百分数的所述铬酸盐即可具备良好的催化活性和稳定性。
进一步优选,所述的催化剂中,所述铬酸盐的重量百分数大于或等于90%。
本发明还提供了一种所述的催化剂在催化氯化氢氧化中的应用,氧气、氯化氢与所述催化剂接触,进行催化氧化反应制得氯气。
作为优选,所述的应用中,将氧气、氯化氢和所述的催化剂接触,进行连续化的管式或床式催化反应,制得氯气。
连续化催化反应优选采用气相固定床连续反应装置。
本发明所述的应用中,例如可在常压下反应。
作为优选,催化氧化反应的温度为360~500℃。
进一步优选,催化氧化反应温度为380~480℃。
更为优选,催化氧化反应温度为400~450℃;最为优选为430℃。
作为优选,氯化氢与氧气的摩尔比为4∶1~1∶4。
进一步优选,氯化氢与氧气的摩尔比为0.5~2。更为优选,氯化氢与氧气的摩尔比为0.5~1。最为优选,氯化氢与氧气的摩尔比为0.5。
更为优选,氯化氢与氧气的摩尔比为0.5~1;反应温度为400~450℃。
氧气与氯化氢混合气体的体积空速500~10000h-1,优选为3000~6000h-1。
本发明所述的催化剂,当粒度在20nm~5μm时,也可采用现有流化床反应装置催化氯化氢氧化制氯气。
本发明还包括一种所述的催化剂的制备方法,所述制备方法采用溶胶-凝胶法或沉淀法:
所述溶胶-凝胶法包括:将M前驱体、Cr前驱体、柠檬酸分散在水中得起始水溶液;随后再与水溶性高分子聚合物混合,水热反应形成凝胶,再经干燥、焙烧、研磨、过筛制得所述催化剂;
所述沉淀法包括:将M前驱体、Cr前驱体分散在水中得混合水溶液,向混合水溶液中投加碱液进行沉淀反应,随后将沉淀产物洗涤、干燥、焙烧、研磨、过筛制得所述催化剂;
所述溶胶-凝胶法和所述沉淀法中,M前驱体为水溶性的钴化合物、锌化合物中的至少一种;Cr前驱体为铬的水溶性化合物。
所述的水溶性钴化合物、锌化合物选自对应金属的氯化物、硝酸盐、醋酸盐等可溶性盐或有机配位化合物。
作为优选,所述钴化合物选自选自氯化钴、硝酸钴、乙酸钴中的至少一种。
作为优选,所述锌化合物选自氯化锌、硝酸锌、乙酸锌中的至少一种。
作为优选,所述铬化合物选自氯化铬、硝酸铬、乙酸铬中的至少一种。
所述的水溶性高分子聚合物优选为聚乙二醇、聚乙烯醇、聚乙烯吡咯烷酮中的至少一种。
所述的溶胶-凝胶法中,先将所述M前驱体、Cr前驱体、柠檬酸溶解在水溶液得起始水溶液,本发明人发现,所述起始水溶液中的M前驱体、Cr前驱体、柠檬酸的浓度对制备的催化剂的活性和稳定性具有一定的影响;作为优选,所述起始水溶液中;Cr的摩尔浓度为0.1~1moL/L。在该起始的铬的浓度下,制备工序效率高,且制得的催化剂的催化性能有所提升。M的摩尔浓度和根据所述的催化剂的M/Cr摩尔比折算。例如,M与Cr的摩尔比为1∶1~1∶4;优选为1∶2~1∶3,最为优选1∶2。
在该优选的Cr的摩尔浓度下,作为优选,所述的起始水溶液中,柠檬酸的浓度为0.1moL/L~2moL/L。进一步优选,溶胶-凝胶法中,所述的起始水溶液中,柠檬酸的浓度为0.2moL/L~1moL/L。
所述的溶胶-凝胶法中,水溶性高分子聚合物的投加量以保证能形成良好凝胶为宜;水溶性高分子聚合物的投加量对制得的催化剂是否有影响,若有,请补充聚合物的投加量,例如,所述的水溶性高分子聚合物的投加量为所述的Cr前驱体重量的0.4~0.8倍。水溶性高分子聚合物起分散剂作用,可采用水溶液形式添加,其质量分数例如为5%~15%。
所述的溶胶-凝胶法中,水浴反应的温度为50~90℃;干燥温度为20~100℃。
所述的溶胶-凝胶法中,焙烧温度为500~1000℃。作为优选,所述的溶胶-凝胶法中,焙烧的温度为600~700℃。更为优选,所述的溶胶-凝胶法中,焙烧的温度为700℃
所述的溶胶-凝胶法制备催化剂过程中,在所述优选温度下,干燥时间优选为2~24小时;焙烧时间优选为2~6小时。
所述的沉淀法中,先将M前驱体、Cr前驱体分散在水溶液得到混合水溶液,其中,混合水溶液中的M前驱体、Cr前驱体浓度对制备的催化剂的催化性能具有一定的影响。本发明人发现,沉淀法中,所述的混合水溶液中,铬的浓度为0.1~1mol/L。在该起始的铬的浓度下,制备的催化剂的比表面的变化不明显,且制备工序效率高,制得的催化剂的催化性能有所提升。M的摩尔浓度和根据所述的催化剂的M/Cr摩尔比折算。例如,M与Cr的摩尔比为1∶1~1∶4;优选为1∶2~1∶3。
所述的沉淀法中,所述的碱液选自碱金属氢氧化物的至少一种的水溶液;或氨水及其水稀释液。
作为优选,所述的碱液选自氨水、氢氧化钠、氢氧化钾中的至少一种的水溶液。
沉淀反应过程中,体系的pH控制在7~8之间。
所述的沉淀法中,沉淀反应的温度为50~90℃,干燥温度为20~100℃。
沉淀法中,焙烧温度为500~1000℃。
作为优选,所述的沉淀法中,焙烧的温度为600~700℃;更为优选,焙烧的温度为700℃。
所述的沉淀法制备催化剂过程中,在所述优选温度下,干燥时间为优选2~24小时;焙烧时间优选2~6小时。
本发明所提供的催化剂,除可用来催化HCl氧化制备氯气外,还可应用于HCl尾气的催化处理领域。
本发明人发现,采用的溶胶凝胶法是制备得到的催化剂的分散性更优,制得的催化剂的稳定性及HCl的催化活性更优于现有其他传统方法,如固相焙烧法。
此外,本发明人还发现,采用的催化剂为溶胶-凝胶法制备的CoCr2O4,在该优选的催化剂下,优选的催化反应温度430℃,氧气与氯化氢的进料摩尔比1∶1。在该优选条件下,催化剂的稳定性高,HCl的催化氧化活性更强。
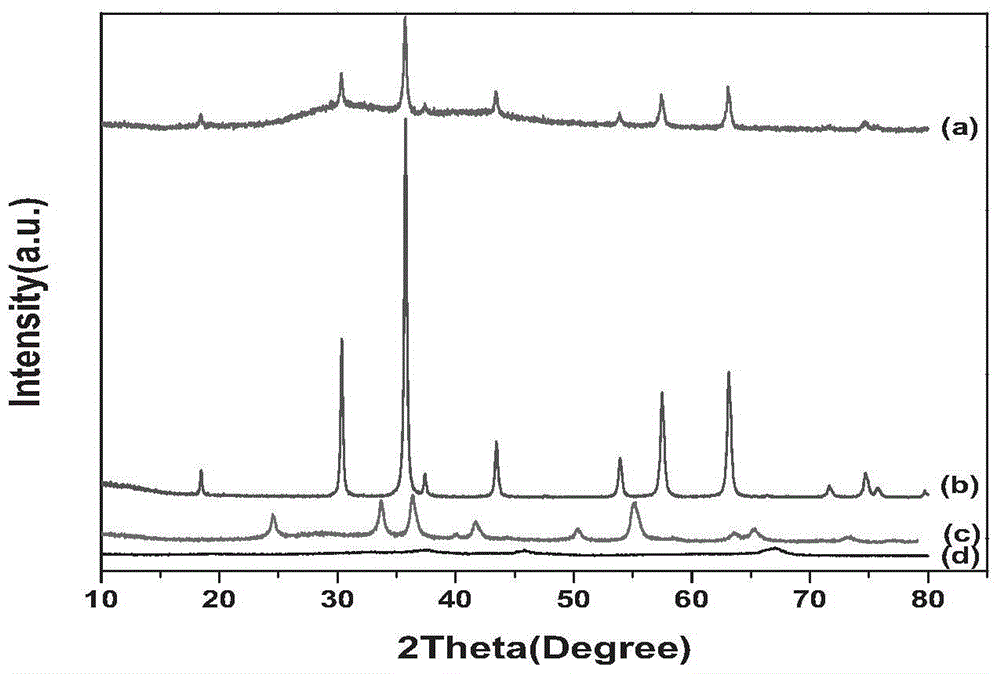
与现有的铜系、铝铬系催化剂相比,催化剂的主要活性成分为铬酸盐MCr2O4(M=Co、Zn)中的一种或几种的混合物,通过XRD图谱可以看出铬酸盐是尖晶石型,其技术优势在于:(1)较高的低温催化活性(如催化反应温度为360~400℃,HCl转化率可以维持在81~90%);(2)反应条件下更高的稳定性;(3)无毒;(4)耐高温(反应温度可高达500℃的条件下,催化剂催化活性也未见失活)。
附图说明
图1为各实施例或对比例制得产品的XRD图谱,其中,(a)CoCr2O4(实施例1),(b)ZnCr2O4(实施例2),(c)Cr2O3(对比例3),(d)Cr2O3/Al2O3(对比例1)
图2为实施例1-2制得的催化剂的SEM照片,其中,图2(a)为实施例1制得的CoCr2O4的SEM图,图2(b)为实施例2制得的ZnCr2O4的SEM图;
图3为各实施例或对比例制得的产品的Cr元素的XPS图谱(a)CoCr2O4(实施例1),(b)ZnCr2O4(实施例2),(c)Cr2O3(对比例3),(d)Cr2O3/Al2O3(对比例1);
图4为各实施例或对比例制得的产品对氯化氢转化率的影响(反应10小时内);其中的CoCr2O4为实施例1制得的产品;CoCr2O4(实施例1),ZnCr2O4(实施例2),Cr2O3(对比例3),Cr2O3/Al2O3(对比例1)
图5为铬酸钴、铬酸锌催化剂的100小时稳定性结果;
图6为反应温度对铬酸钴催化剂活性的影响;
图7为氯化氢和氧气进料比对铬酸钴催化剂活性的影响。
具体实施方式:
【实施例1】溶胶-凝胶法铬酸钴(CoCr2O4)催化剂(钴铬摩尔比1∶2)
(a)将1.82g的六水合硝酸钴,5.0克九水硝酸铬,4.0g的一水柠檬酸,加入25mL水的蒸馏水配成溶液(起始水溶液);
(b)把(a)得到的溶液在80℃下水浴加热搅拌,加入分散剂2~3g聚乙二醇(400),当水分快要蒸发干并成粘稠的凝胶,放到80℃下烘干2h,并经700℃煅烧(焙烧)2h;
(c)取出煅烧好的催化剂,经研钵研磨后取40~60目的催化剂进行氯化氢催化反应。
经XRD图谱分析,本实施例制得的CoCr2O4的XRD图谱见图1的(a)谱线,其与标准CoCr2O4图谱(JCPDS 22-1084)吻合。
本实施例制得的CoCr2O4的SEM见图2的(a)部分。
本实施例制得的CoCr2O4的XPS见图3的(a)谱线。
经元素分析得出:催化剂中钴占催化剂重量的26.4%,铬占催化剂重量的46.4%,铬酸钴占催化剂含量99.4%。催化剂比表面积为29.5m2/g,XRD计算晶体粒径大小为18nm。
【实施例2】溶胶-凝胶法铬酸锌(ZnCr2O4)催化剂(锌铬摩尔比1∶2)
(a)将1.86g的六水合硝酸锌,5.0克九水硝酸铬,4.0g的一水柠檬酸,加入到25mL的蒸馏水配成溶液;
(b)把(a)得到的溶液在80℃下水浴加热搅拌,加入分散剂2~3g聚乙二醇(400),当水分快要蒸发干并成粘稠的凝胶,放到80℃下烘干2h,并经700℃煅烧(焙烧)2h;
(c)取出煅烧好的催化剂,经研钵研磨后取40~60目的催化剂进行氯化氢催化反应。
经XRD图谱分析,本实施例制得的ZnCr2O4的XRD图谱见图1的(b)谱线,其与标准ZnCr2O4图谱(JCPDS 22-1107)吻合。
本实施例制得的ZnCr2O4的SEM见图2的(b)部分。
本实施例制得的ZnCr2O4的XPS见图3的(b)谱线。
经元素分析得出:催化剂中锌占催化剂重量的25.3%,铬占催化剂重量的45.7%,铬酸锌占催化剂含量94.6%。催化剂比表面积为24.6m2/g,XRD计算晶体粒径大小为27nm。。
【实施例3】沉淀法铬酸钴(CoCr2O4)催化剂(钴铬摩尔比1∶2)
(a)将1.82g的六水合硝酸钴,5.0克九水硝酸铬,加入到25mL的蒸馏水配成溶液(混合水溶液);
(b)把(a)得到的溶液在80℃下水浴加热,然后加入氨水调节到pH=7左右,有大量的沉淀生成。停止搅拌继续水浴加热,陈化一段时间让生成的沉淀不断长大。通过过滤洗涤后,放到80℃下烘干2h,并经700℃煅烧(焙烧)2h;
(c)取出煅烧好的催化剂,经研钵研磨后取40~60的催化剂进行氯化氢催化反应。
经元素分析得出:催化剂中钴占催化剂重量的25.8%,铬占催化剂重量的44.2%,铬酸钴占催化剂含量90.4%,催化剂比表面积为19.6m2/g,经XRD计算晶体粒径大小为47nm。
【实施例4】溶胶-凝胶法铬酸钴(CoCr2O4)催化剂(钴铬摩尔比1∶1)
(a)将3.64g的六水合硝酸钴,5.0克九水硝酸铬,4.0g的一水柠檬酸,加入25mL的蒸馏水配成溶液;
(b)把(a)得到的溶液在80℃下水浴加热搅拌,加入分散剂2~3g聚乙二醇(400),当水分快要蒸发干并成粘稠的凝胶,放到80℃下烘干2h,并经700℃煅烧(焙烧)2h;
(c)取出煅烧好的催化剂,经研钵研磨后取40~60目的催化剂进行氯化氢催化反应。
经元素分析得出:经元素分析得出:催化剂中钴占催化剂重量的38.4%,铬占催化剂重量的32.4%,铬酸钴占催化剂含量75%,催化剂比表面积为24.3m2/g。
【实施例5】溶胶-凝胶法铬酸钴(CoCr204)催化剂(钴铬摩尔比1∶2,和实施例1相比,一水柠檬酸的投料量影响(一水柠檬酸的加入量减小到一半))
(a)将1.82g的六水合硝酸钴,5.0克九水硝酸铬,2.0g的一水柠檬酸,加入25mL的蒸馏水配成溶液;
(b)把(a)得到的溶液在80℃下水浴加热搅拌,加入分散剂2~3g聚乙二醇(400),当水分快要蒸发干并成粘稠的凝胶,放到80℃下烘干2h,并经700℃煅烧(焙烧)2h;
(c)取出煅烧好的催化剂,经研钵研磨后取40~60目的催化剂进行氯化氢催化反应。
经元素分析得出:催化剂中钴占催化剂重量的26.2%,铬占催化剂重量的46.7%,铬酸钴占催化剂含量92.4%。催化剂比表面积为25.1m2/g。
【实施例6】溶胶-凝胶法铬酸钴(CoCr2O4)催化剂(钴铬摩尔比1∶2,和实施例1相比,煅烧温度升高到900℃)
(a)将1.82g的六水合硝酸钴,5.0克九水硝酸铬,4.0g的一水柠檬酸,加入25mL的蒸馏水配成溶液;
(b)把(a)得到的溶液在80℃下水浴加热搅拌,加入分散剂2~3g聚乙二醇(400),当水分快要蒸发干并成粘稠的凝胶,放到80℃下烘干2h,并经900℃煅烧(焙烧)2h;
(c)取出煅烧好的催化剂,经研钵研磨后取40~60目的催化剂进行氯化氢催化反应。
经元素分析得出:催化剂中钴占催化剂重量的26.7%,铬占催化剂重量的46.3%,铬酸钴占催化剂含量99.2%。催化剂比表面积为18.2m2/g。
【对比例1】氧化铬/活性氧化铝浸渍催化剂
(a)将5.0g的九水合硝酸铬,加入到25mL的蒸馏水中配成溶液,向所述溶液中加入40~60目活性Al2O3载体并浸渍12小时;
(b)滤去所述溶液得到浸渍过的载体,在80℃下烘干2h,并经700℃煅烧2h。得到具有催化活性的催化剂。
本对比例制得的氧化铬/活性氧化铝的XRD图谱见图1的(d)谱线。
本对比例制得的氧化铬/活性氧化铝的XPS见图3的(d)谱线。
经元素分析得出:催化剂中铬占催化剂重量的7.5%。催化剂比表面积为205.4m2/g。
【对比例2】氧化铬/二氧化钛浸渍催化剂
(a)将5.0g的九水合硝酸铬,加入25mL的蒸馏水中配成溶液,向所述溶液中加入40~60目活性TiO2载体并浸渍12小时;
(b)滤去所述溶液得到浸渍过的载体,在80℃下烘干2h,并经700℃煅烧2h。得到具有催化活性的催化剂。
经元素分析得出:催化剂中铬占催化剂重量的8.3%,催化剂比表面积为155.3m2/g。
【对比例3】溶胶-凝胶法氧化铬催化剂
(a)将5.0克九水硝酸铬,4.0g的一水柠檬酸,加入25mL的蒸馏水配成溶液;
(b)把溶液在80℃下水浴加热搅拌,加入分散剂2~3g聚乙二醇(400),当水分快要蒸发干并成粘稠的凝胶,放到80℃下烘干2h,并经700℃煅烧2h;
(c)取出煅烧好的催化剂,经研钵研磨后取40~60的催化剂进行氯化氢催化反应。
本对比例制得的氧化铬的XRD图谱见图1的(c)谱线。
本对比例制得的氧化铬的XPS见图3的(c)谱线。
经元素分析得出:催化剂中铬占催化剂重量含量67%。催化剂比表面积为13.7m2/g。
【实施例7】催化剂性能评价
对上述实施例1~6的催化剂和对比例1-3制得的对比催化剂在固定床反应器中进行氯化氢催化氧化制氯气的性能评价。反应管中加入0.7g催化剂,以5℃/分钟的升温速率从室温加热到430℃,通入氯化氢和氧气的混合气体,氯化氢流量为10mL/min,氧气流量为10mL/min,载气氮气流量150mL/min时,反应2小时,用100mL浓度为10%的KI溶液吸收3分钟,用已标定的Na2S2O3溶液滴定生成的Cl2。计算HCl的转化率,并以单位质量催化剂单位时间内生成的氯气量作为催化剂的活性。
本发明制备的催化剂的性能比较结果如表1和图4所示。
表1催化剂氯化氢催化氧化制氯气性能比较
对比例4*为专利EP0184413买施例1的数据,对比例5**为专利US5707919实施例3的数据,对比例6***为专利CN104785271A实施例1的数据。
从表1、图4可以看出,(1)实施例1~2表现良好的活性与稳定性。其活性高于其他几个相关专利报导的数据。(2)对比例1,稳定了4小时后开始下降,说明负载型的三氧化二铬与载体之间的作用力不够,以至于一段时间后三氧化二铬开始流失。(3)对比例3,纯粹的三氧化二铬催化剂稳定性和活性都很低,与文献报道吻合。(4)最优催化剂是实施例1的铬酸钴催化剂。
【实施例8】CoCr2O4和ZnCr2O4的催化稳定性测试
以实施例1中催化剂CoCr2O4和和实施例2的ZnCr2O4为例,研究尖晶石型催化剂的催化稳定性。测试条件为实施例7所述,测试结果在图5中显示。如图5所述,由上而下分别为CoCr2O4、ZnCr2O4的测定结果。(实验条件等同于实施例7:催化剂为0.7g,实施例1和实施例2的催化剂,反应温度为430℃,HCl=10mL/min,O2=10mL/min,N2=150mL/min。)
由图5可以看出,(1)铬酸钴、铬酸锌的稳定性和催化活性都挺高的。(2)反应100小时后,铬酸钴的活性下降了2%,铬酸锌的活性下降了5%,说明铬酸钴稳定性更好。
【实施例9】反应温度对催化活性影响
以实施例1中催化剂CoCr2O4为例,研究尖晶石型催化剂在不同的反应温度下,氯化氢的转化率的变化。测试条件参见实施例7,仅反应温度从320℃到500℃变化。测试结果在图6中显示。(实验条件:催化剂为0.7g,实施例1催化剂,HCl=10mL/min,O2=10mL/min,N2=150mL/min。改变反应温度T=320~500℃。)
由图6可以看出,当温度在320℃时,反应活性还比较低,仅仅45%;但是当温度升高到360℃时,转化率迅速提高到81%;随着温度继续上升,转化率缓慢上升,在430℃达到最大93%,接近反应平衡。由此看出当反应温度360~400℃,催化剂可以具有81~93%的氯化氢转化率。这个性能高于文献中报导的同温度下得性能。说明本专利的催化剂具有较好的低温活性。当温度继续升高,转化率开始下降。这是因为本反应是放热反应,温度越高反应开始向左移动。
【实施例10】氯化氢与氧气比例对CoCr2O4催化活性影响
以实施例1中催化剂CoCr2O4为例,研究尖晶石型催化剂在不同的进料比下,氯化氢的转化率的变化。测试条件为实施例7,区别在于进料比变化HCl/O2=4~0.5。(实验条件:催化剂为0.7g,实施例1催化剂,反应温度为430℃,HCl=10mL/min,N2=150mL/min。改变氧气的流量分别为2.5mL/min、5mL/min、10mL/min、20mL/min。)测试结果在图7中显示。
由图7可以看出,当氯化氢和氧气的进料摩尔比为4∶1时,转化率小于50%;当氯化氢和氧气的进料摩尔比为2∶1时,转化率迅速提高到84%;当氯化氢和氧气的进料摩尔比为1∶1时,转化率达到最大93%;当氯化氢和氧气的流量比继续增大,转化率不再增加。
- 还没有人留言评论。精彩留言会获得点赞!